

m6A modification of mutant huntingtin RNA promotes the biogenesis of pathogenic huntingtin transcripts
- PMID: 39394467
- PMCID: PMC11549361
- DOI: 10.1038/s44319-024-00283-7
m6A modification of mutant huntingtin RNA promotes the biogenesis of pathogenic huntingtin transcripts
Abstract
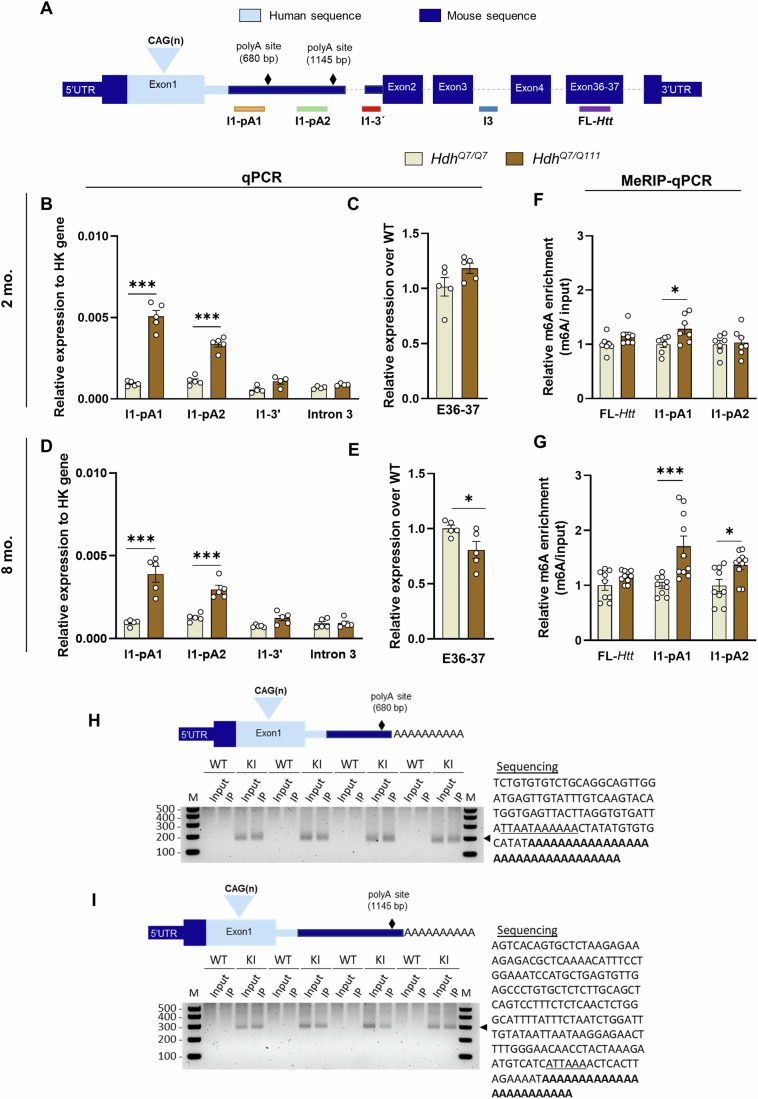
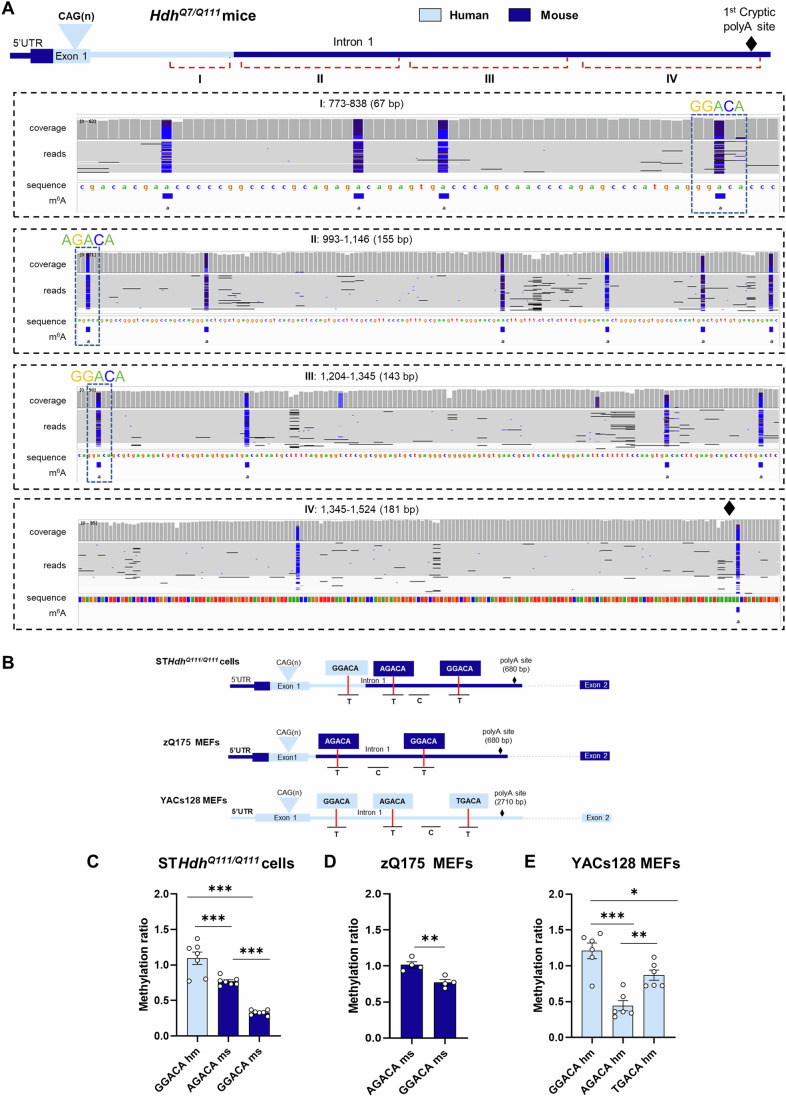
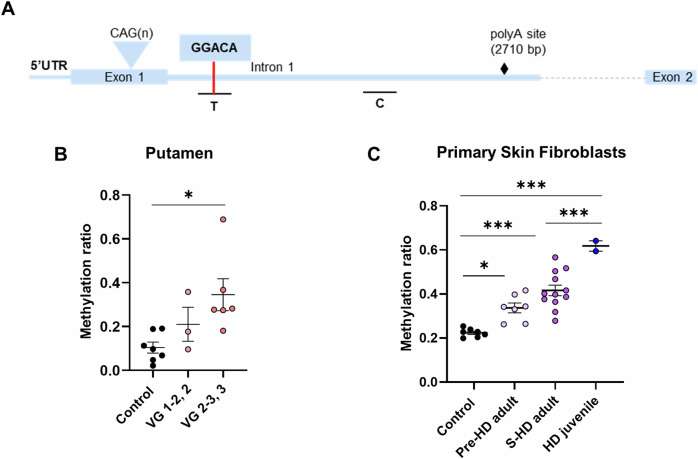
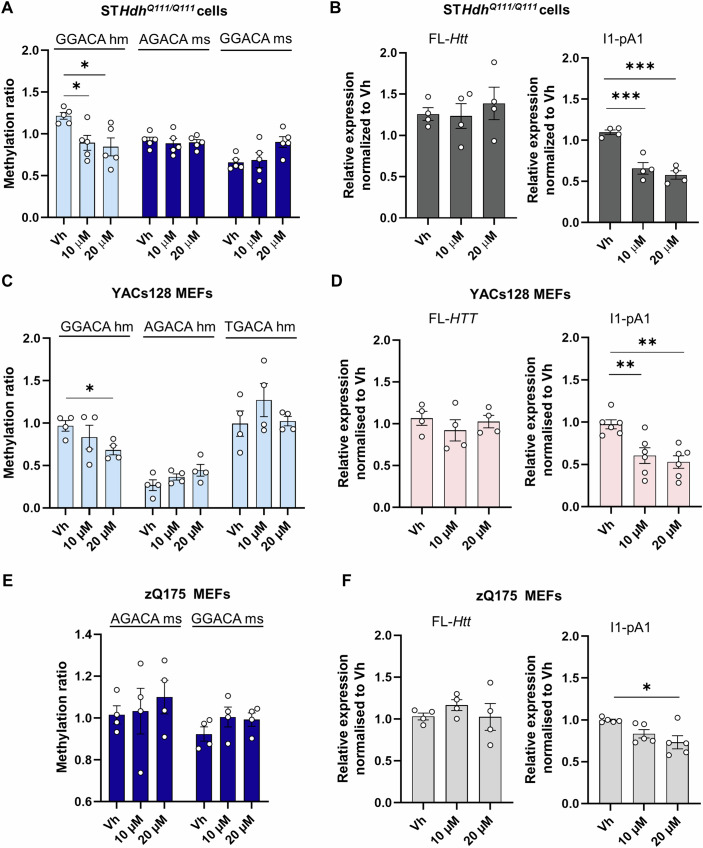
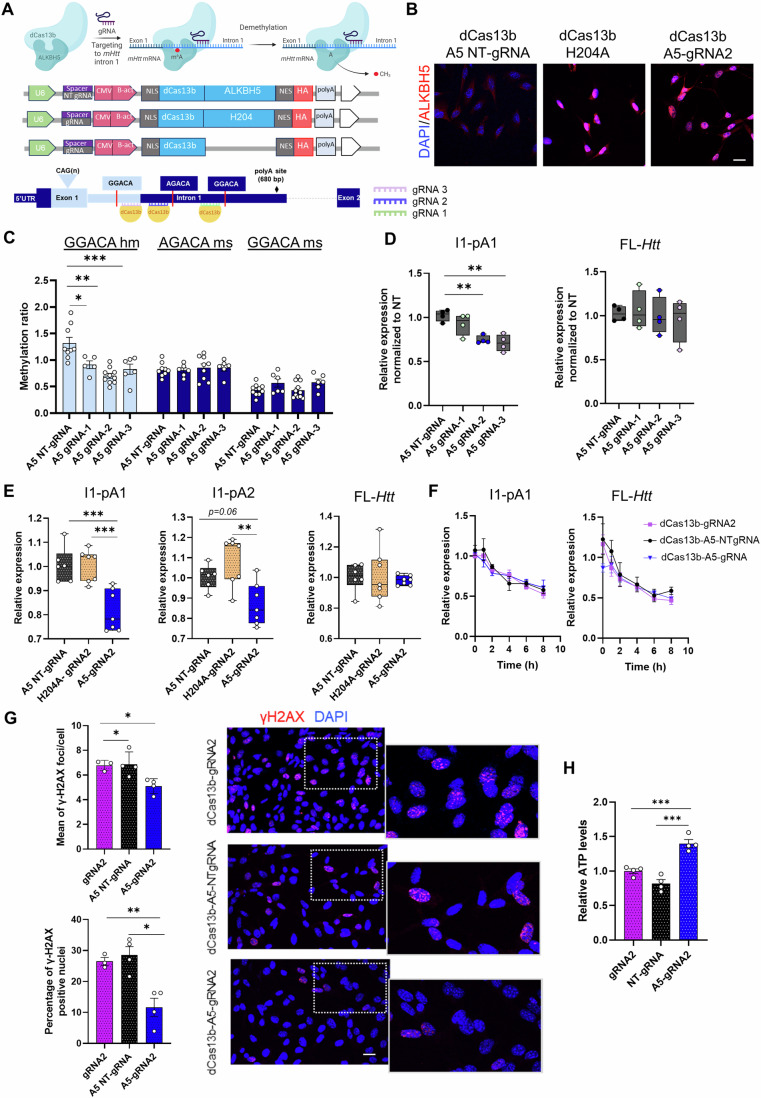
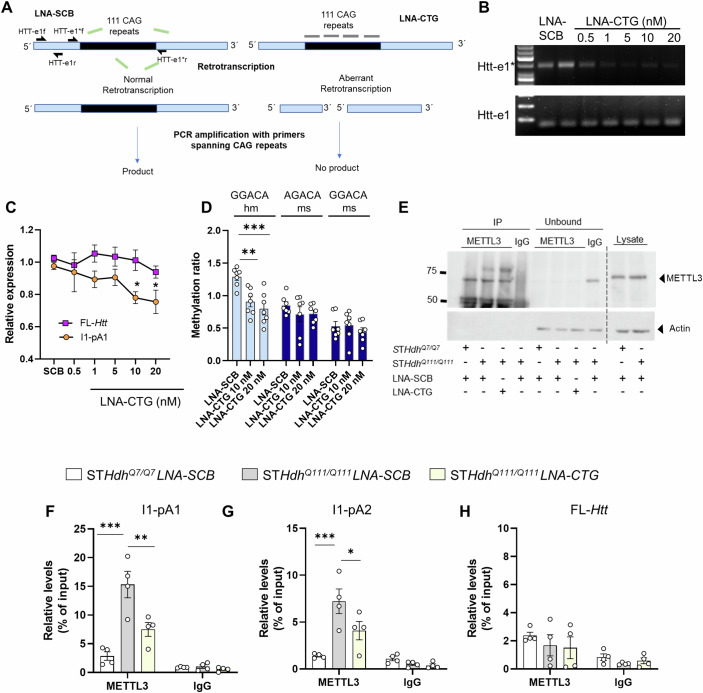
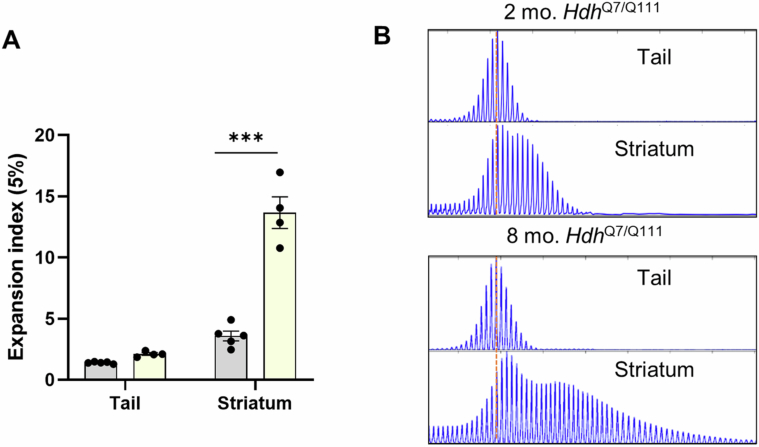
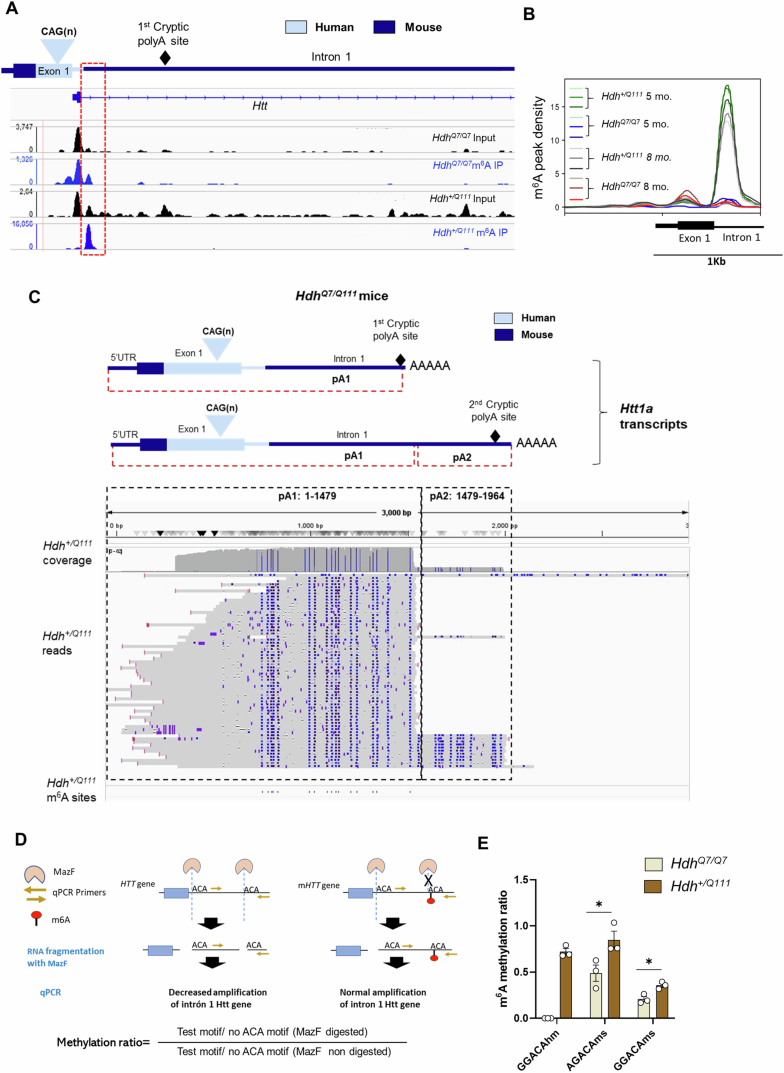
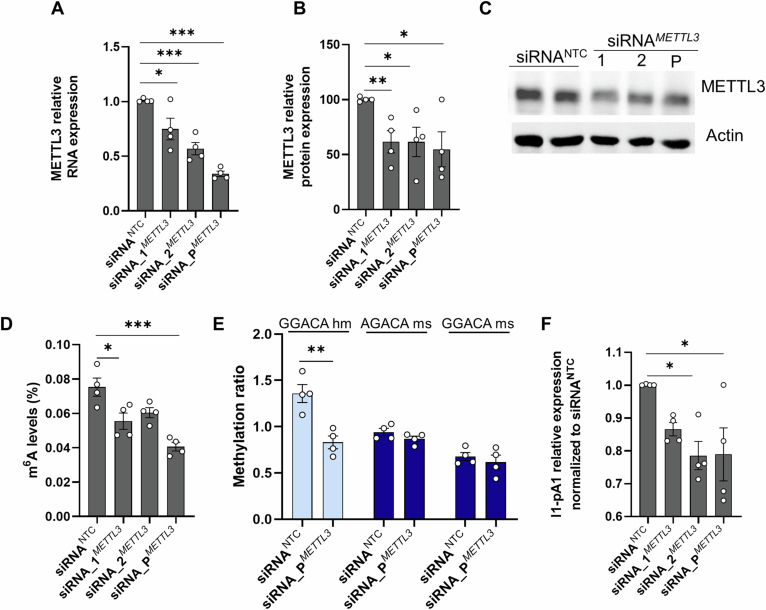
In Huntington's disease (HD), aberrant processing of huntingtin (HTT) mRNA produces HTT1a transcripts that encode the pathogenic HTT exon 1 protein. The mechanisms behind HTT1a production are not fully understood. Considering the role of m6A in RNA processing and splicing, we investigated its involvement in HTT1a generation. Here, we show that m6A methylation is increased before the cryptic poly(A) sites (IpA1 and IpA2) within the huntingtin RNA in the striatum of Hdh+/Q111 mice and human HD samples. We further assessed m6A's role in mutant Htt mRNA processing by pharmacological inhibition and knockdown of METTL3, as well as targeted demethylation of Htt intron 1 using a dCas13-ALKBH5 system in HD mouse cells. Our data reveal that Htt1a transcript levels are regulated by both METTL3 and the methylation status of Htt intron 1. They also show that m6A methylation in intron 1 depends on expanded CAG repeats. Our findings highlight a potential role for m6A in aberrant splicing of Htt mRNA.
Keywords: HTT1a; Huntington’s Disease; RNA Editing; Splicing; m6A.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
MeSH terms
Substances
Grants and funding
- RTI2018-094374-B100/Ministerio de Ciencia, Innovación y Universidades (MCIU)
- PID2020-113953RB-I00/Ministerio de Ciencia, Innovación y Universidades (MCIU)
- PID2020-116474RB-100/Ministerio de Ciencia, Innovación y Universidades (MCIU)
- RE2021-097199/Ministerio de Ciencia, Innovación y Universidades (MCIU)
- R01 NS126420/NS/NINDS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
