

A comprehensive proteogenomic pipeline for neoantigen discovery to advance personalized cancer immunotherapy
- PMID: 39394480
- PMCID: PMC12339364
- DOI: 10.1038/s41587-024-02420-y
A comprehensive proteogenomic pipeline for neoantigen discovery to advance personalized cancer immunotherapy
Abstract
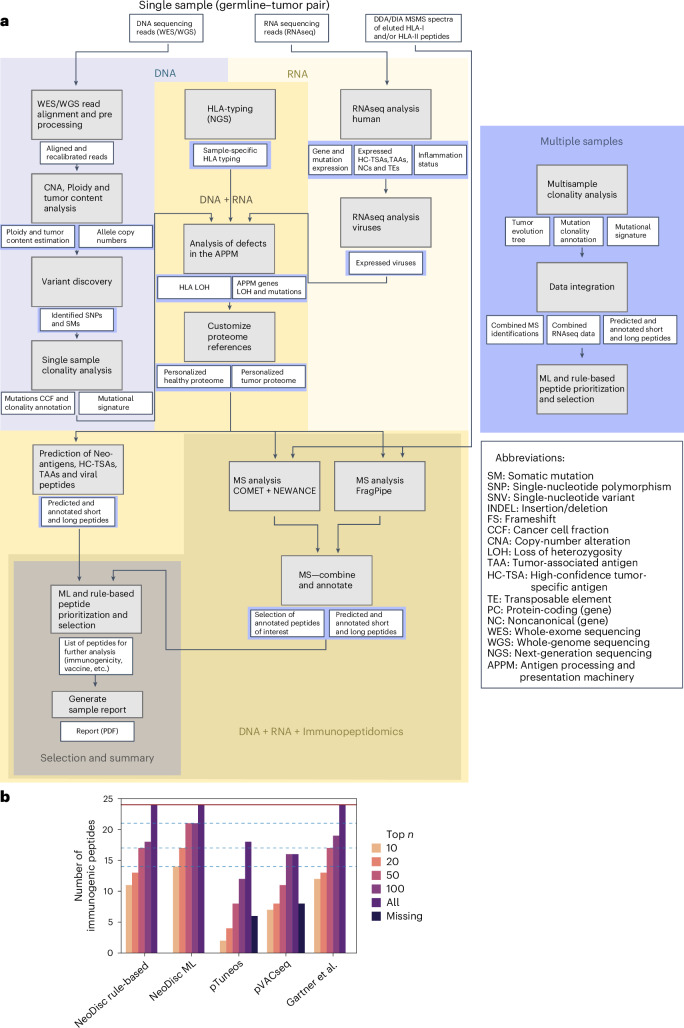
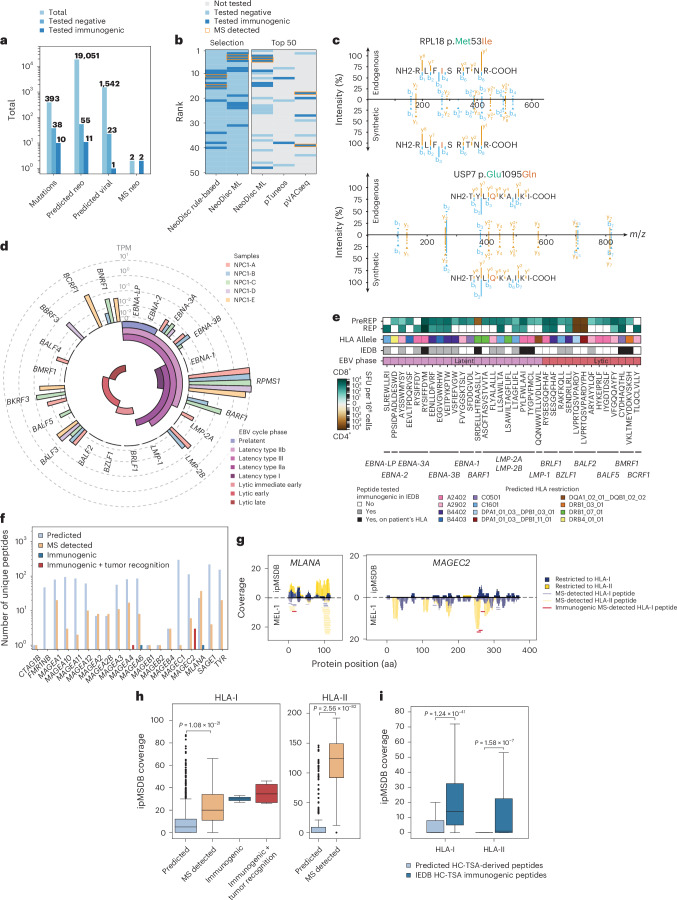
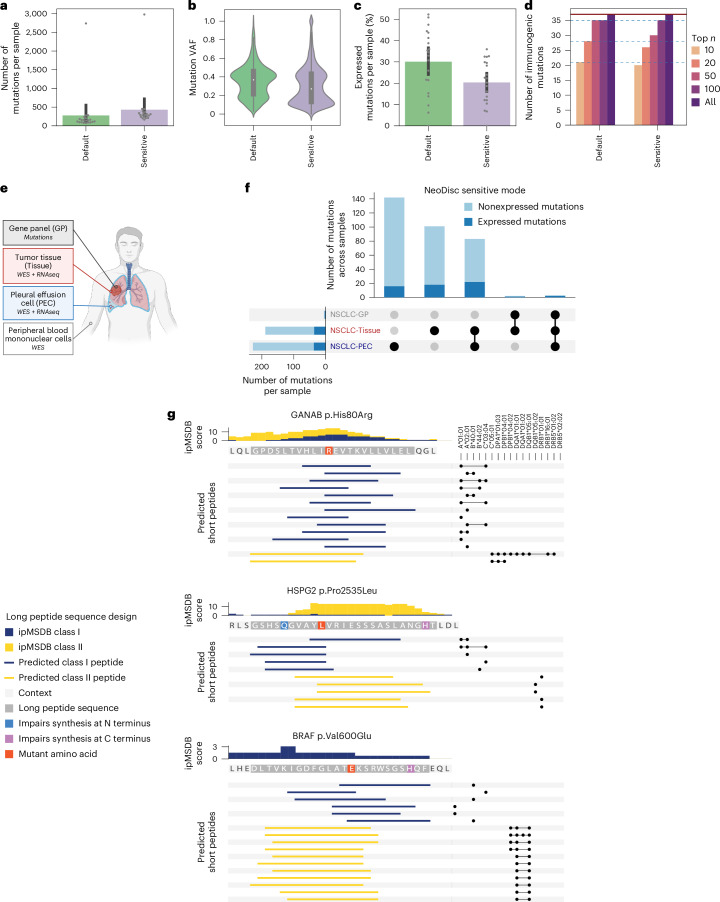
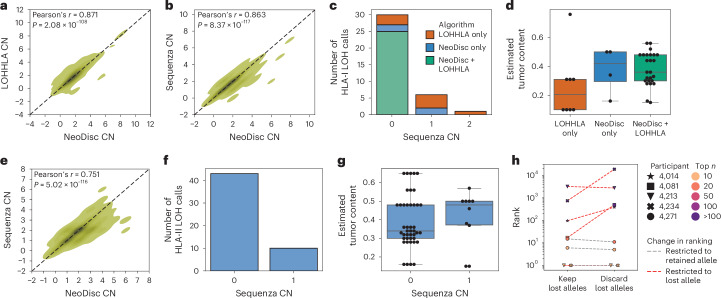
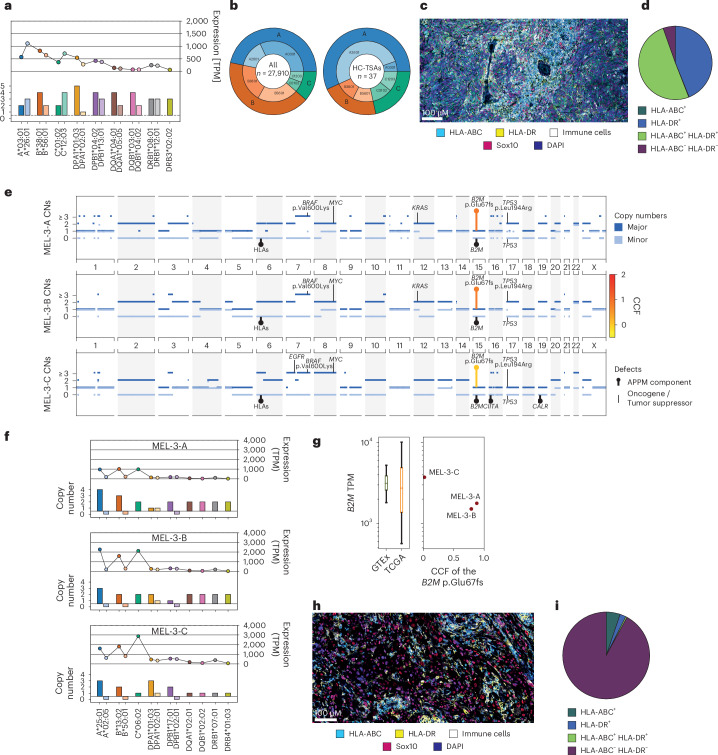
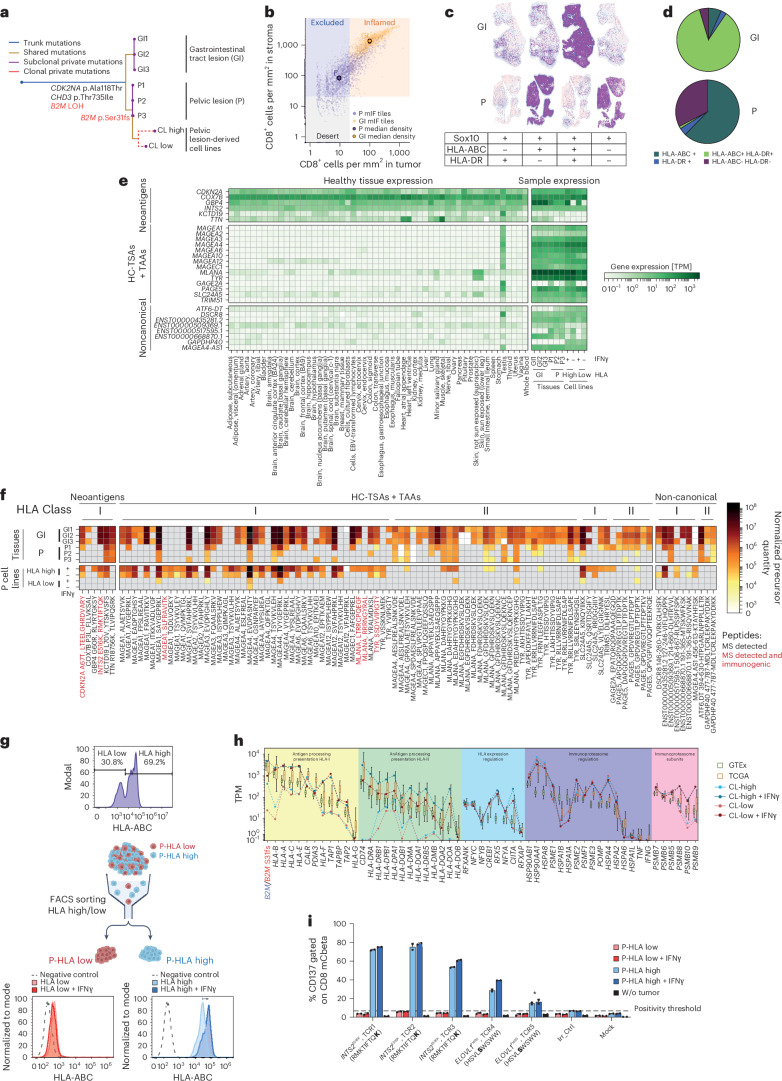
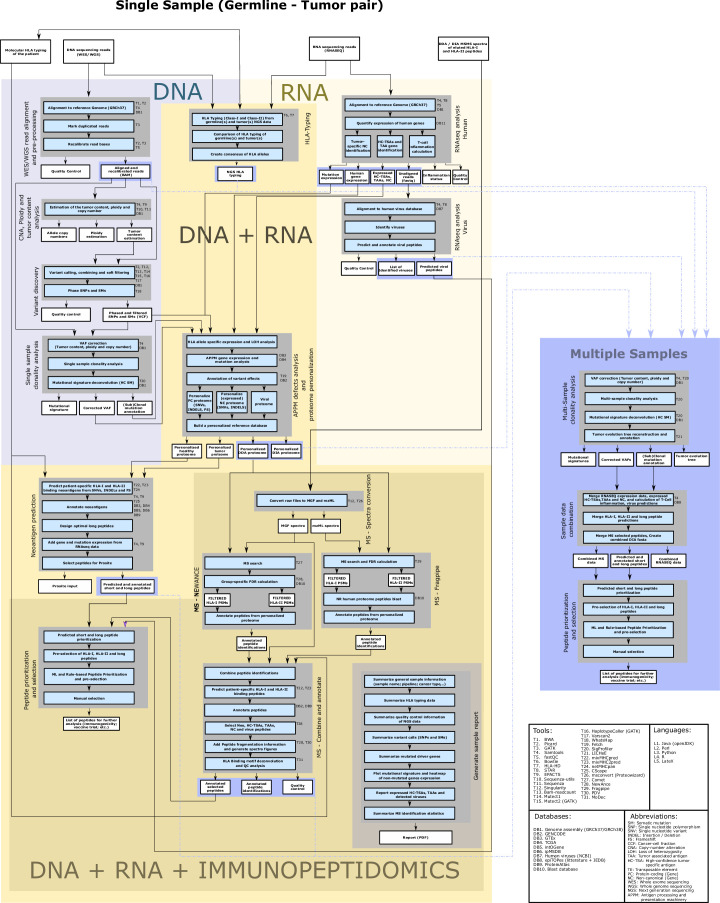
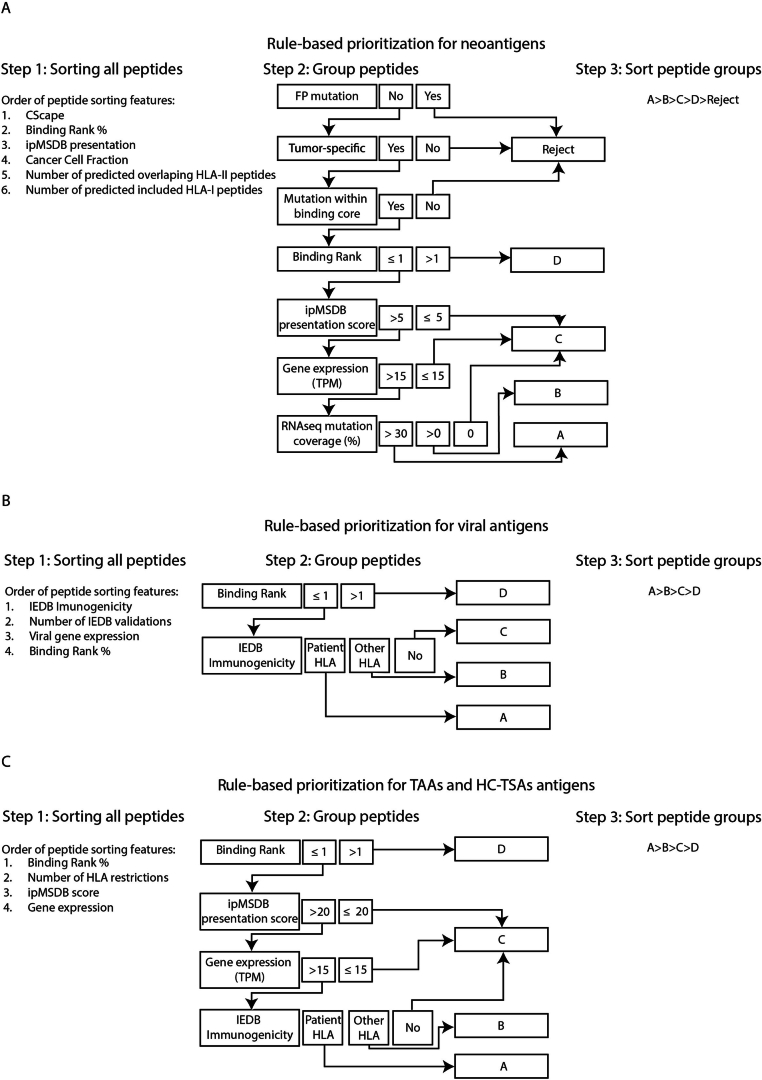
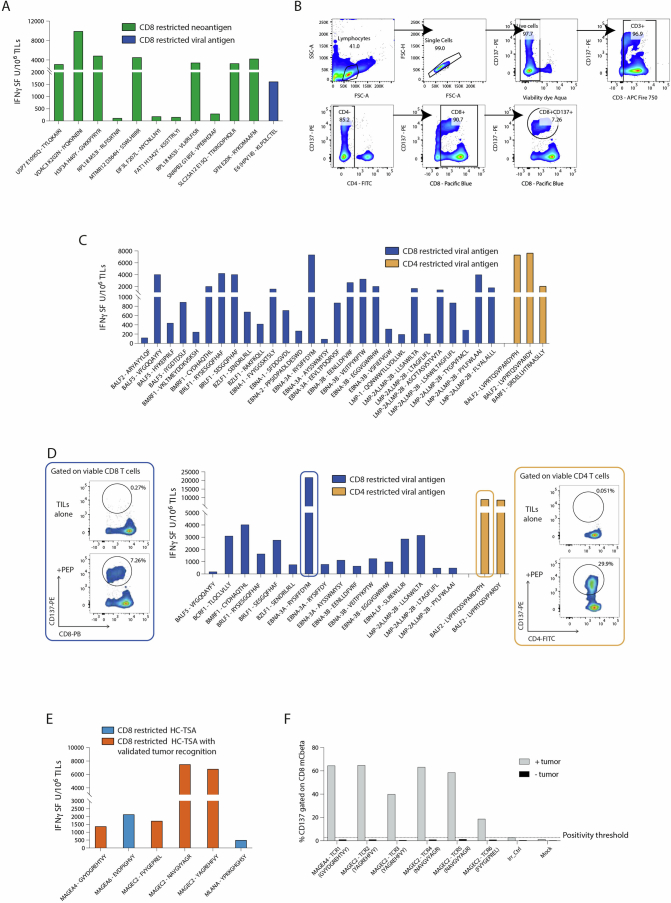
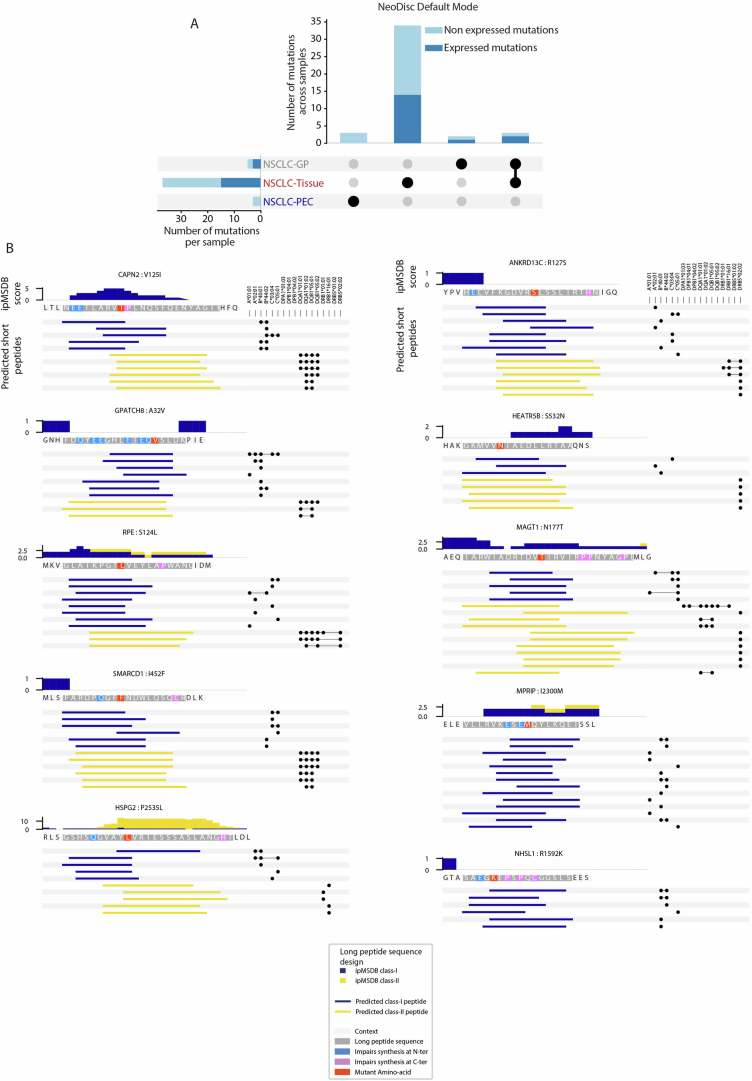
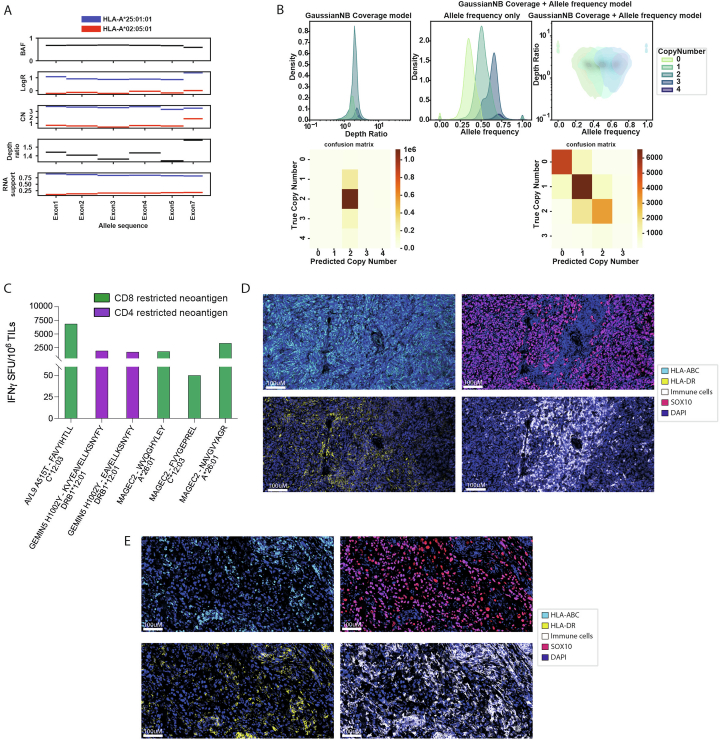
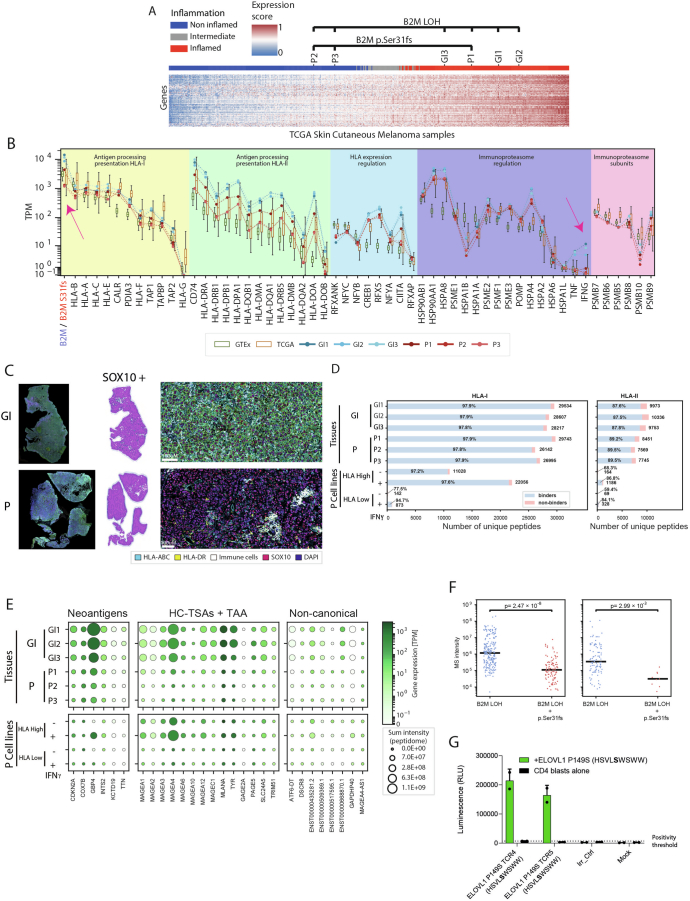
The accurate identification and prioritization of antigenic peptides is crucial for the development of personalized cancer immunotherapies. Publicly available pipelines to predict clinical neoantigens do not allow direct integration of mass spectrometry immunopeptidomics data, which can uncover antigenic peptides derived from various canonical and noncanonical sources. To address this, we present an end-to-end clinical proteogenomic pipeline, called NeoDisc, that combines state-of-the-art publicly available and in-house software for immunopeptidomics, genomics and transcriptomics with in silico tools for the identification, prediction and prioritization of tumor-specific and immunogenic antigens from multiple sources, including neoantigens, viral antigens, high-confidence tumor-specific antigens and tumor-specific noncanonical antigens. We demonstrate the superiority of NeoDisc in accurately prioritizing immunogenic neoantigens over recent prioritization pipelines. We showcase the various features offered by NeoDisc that enable both rule-based and machine-learning approaches for personalized antigen discovery and neoantigen cancer vaccine design. Additionally, we demonstrate how NeoDisc's multiomics integration identifies defects in the cellular antigen presentation machinery, which influence the heterogeneous tumor antigenic landscape.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: G.C. has received honoraria from Bristol-Myers Squibb. CHUV has received honoraria for advisory services provided by G.C. to Iovance and EVIR. G.C. has received royalties from the University of Pennsylvania for CAR T cell therapy licensed to Novartis and Tmunity Therapeutics. G.C., A.H., M.A., S.B., F.H. and M.B.-S. have received royalties from the Ludwig Institute for Cancer Research, UNIL and CHUV for NeoTIL intellectual property previously licensed to Tigen Pharma. S.B., G.C. and A.H. are inventors in technologies related to T cell expansion and engineering for T cell therapy. F.H., M.M. and M.B.-S. are inventors on a patent application related to ML prioritization of neoantigens. F.H. and M.B.-S. are inventors on a patent application filed under certain subject matters disclosed herein. D.G. has received honoraria for consultations from CeCaVa and Gnubiotics. The other authors declare no competing interests.
Figures
References
-
- Chong, C., Coukos, G. & Bassani-Sternberg, M. Identification of tumor antigens with immunopeptidomics. Nat. Biotechnol.40, 175–188 (2022). - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
