

Immunometabolic changes and potential biomarkers in CFS peripheral immune cells revealed by single-cell RNA sequencing
- PMID: 39394558
- PMCID: PMC11468054
- DOI: 10.1186/s12967-024-05710-w
Immunometabolic changes and potential biomarkers in CFS peripheral immune cells revealed by single-cell RNA sequencing
Abstract
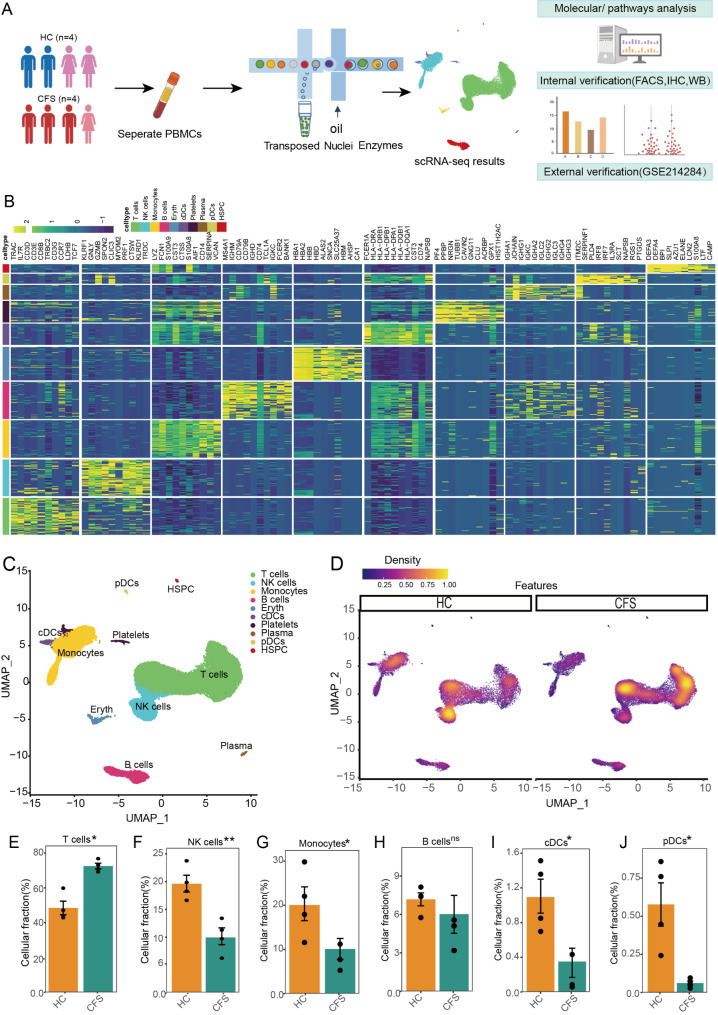
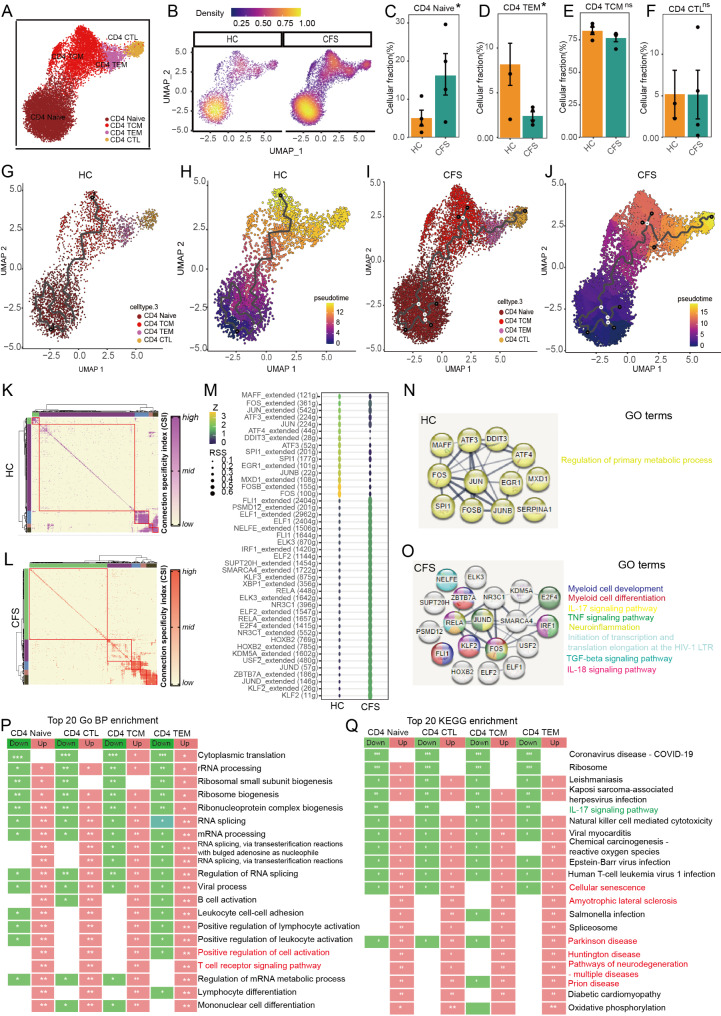
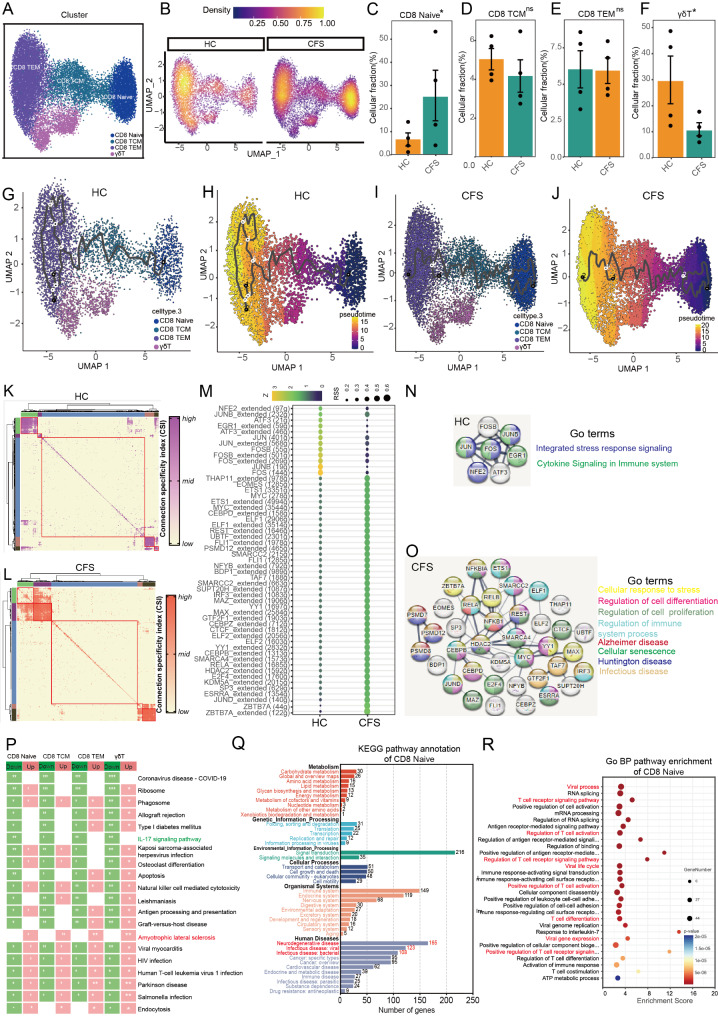
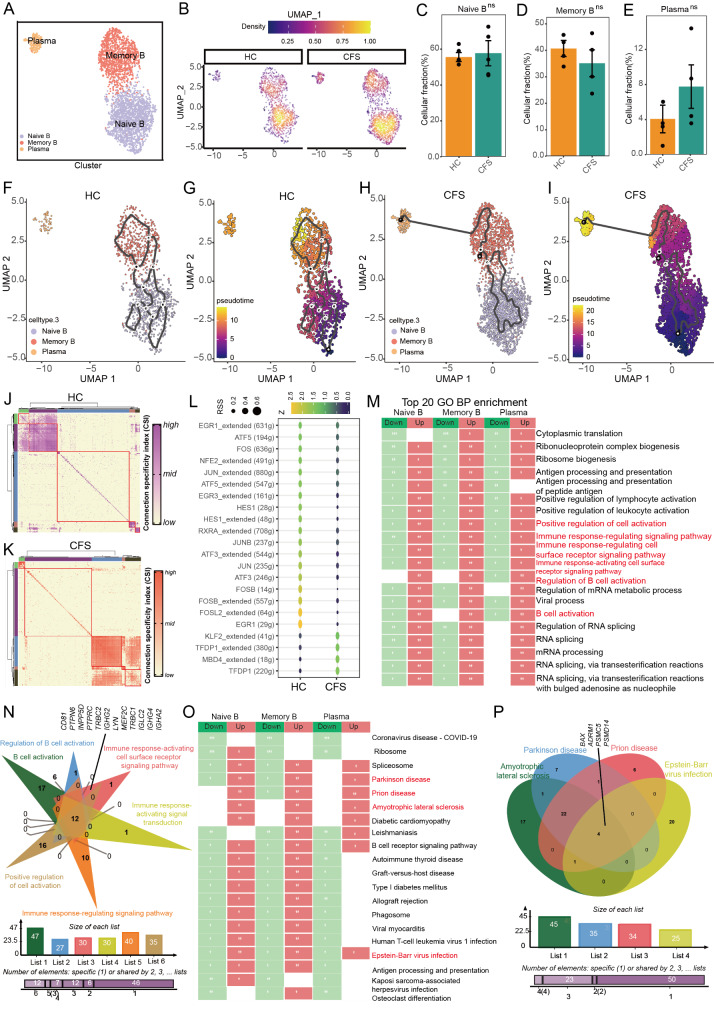
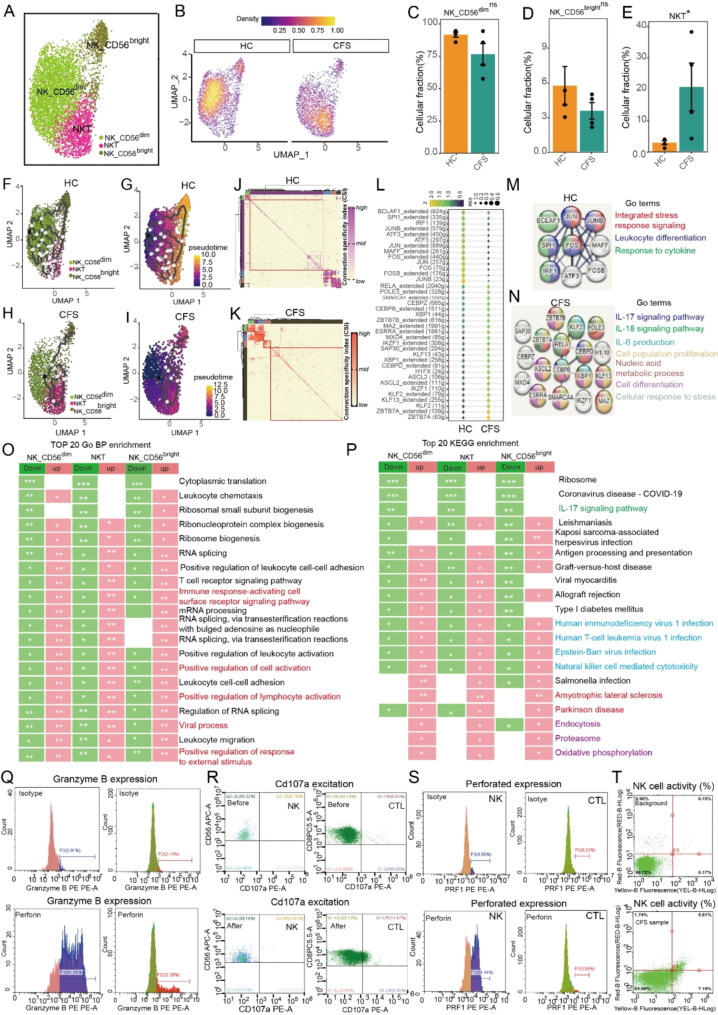
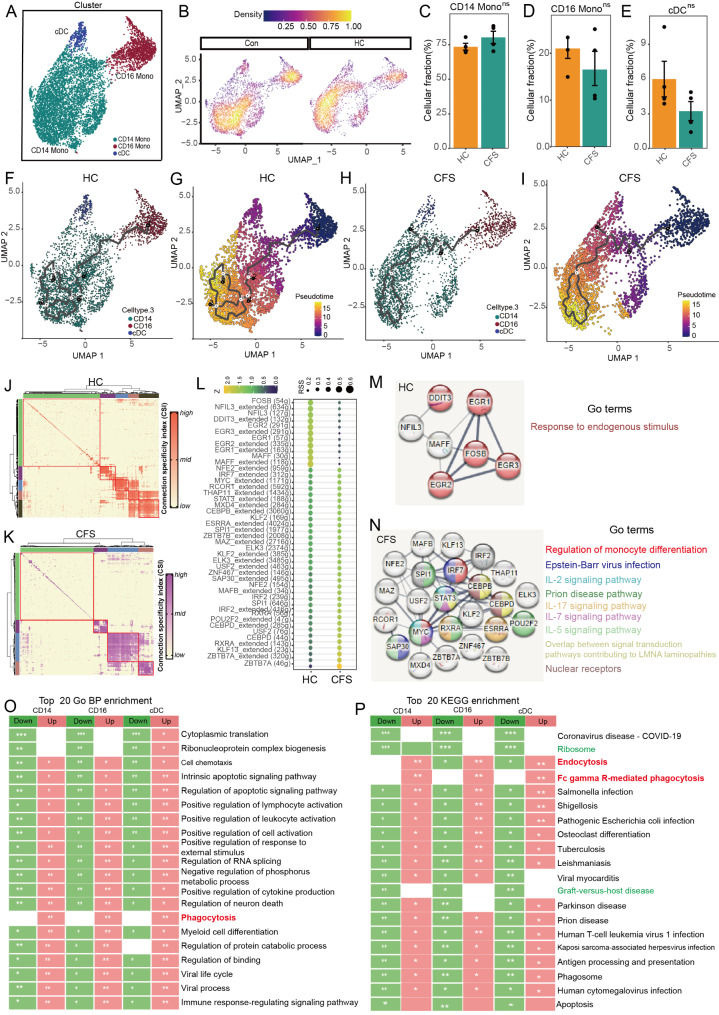
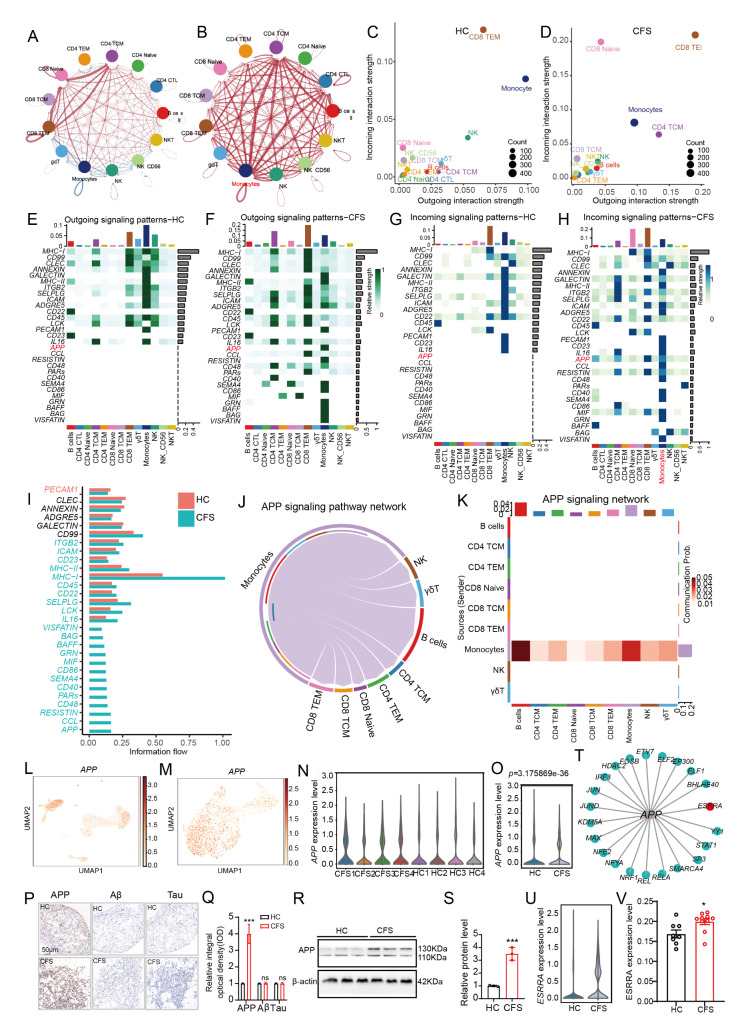
The pathogenesis of Myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) remains unclear, though increasing evidence suggests inflammatory processes play key roles. In this study, single-cell RNA sequencing (scRNA-seq) of peripheral blood mononuclear cells (PBMCs) was used to decipher the immunometabolic profile in 4 ME/CFS patients and 4 heathy controls. We analyzed changes in the composition of major PBMC subpopulations and observed an increased frequency of total T cells and a significant reduction in NKs, monocytes, cDCs and pDCs. Further investigation revealed even more complex changes in the proportions of cell subpopulations within each subpopulation. Gene expression patterns revealed upregulated transcription factors related to immune regulation, as well as genes associated with viral infections and neurodegenerative diseases.CD4+ and CD8+ T cells in ME/CFS patients show different differentiation states and altered trajectories, indicating a possible suppression of differentiation. Memory B cells in ME/CFS patients are found early in the pseudotime, indicating a unique subtype specific to ME/CFS, with increased differentiation to plasma cells suggesting B cell overactivity. NK cells in ME/CFS patients exhibit reduced cytotoxicity and impaired responses, with reduced expression of perforin and CD107a upon stimulation. Pseudotime analysis showed abnormal development of adaptive immune cells and an enhanced cell-cell communication network converging on monocytes in particular. Our analysis also identified the estrogen-related receptor alpha (ESRRA)-APP-CD74 signaling pathway as a potential biomarker for ME/CFS in peripheral blood. In addition, data from the GSE214284 database confirmed higher ESRRA expression in the monocyte cell types of male ME/CFS patients. These results suggest a link between immune and neurological symptoms. The results support a disease model of immune dysfunction ranging from autoimmunity to immunodeficiency and point to amyloidotic neurodegenerative signaling pathways in the pathogenesis of ME/CFS. While the study provides important insights, limitations include the modest sample size and the evaluation of peripheral blood only. These findings highlight potential targets for diagnostic biomarkers and therapeutic interventions. Further research is needed to validate these biomarkers and explore their clinical applications in managing ME/CFS.
Keywords: Biomarkers; Immune dysregulation; Immunometabolism; Myalgic encephalomyelitis/Chronic fatigue syndrome (ME/CFS); Peripheral blood mononuclear cells (PBMCs); Single-cell RNA sequencing (scRNA-seq).
© 2024. The Author(s).
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Figures
Similar articles
-
MicroRNAs hsa-miR-99b, hsa-miR-330, hsa-miR-126 and hsa-miR-30c: Potential Diagnostic Biomarkers in Natural Killer (NK) Cells of Patients with Chronic Fatigue Syndrome (CFS)/ Myalgic Encephalomyelitis (ME).PLoS One. 2016 Mar 11;11(3):e0150904. doi: 10.1371/journal.pone.0150904. eCollection 2016. PLoS One. 2016. PMID: 26967895 Free PMC article.
-
Validation of impaired Transient Receptor Potential Melastatin 3 ion channel activity in natural killer cells from Chronic Fatigue Syndrome/ Myalgic Encephalomyelitis patients.Mol Med. 2019 Apr 23;25(1):14. doi: 10.1186/s10020-019-0083-4. Mol Med. 2019. PMID: 31014226 Free PMC article.
-
Cellular Immune Function in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS).Front Immunol. 2019 Apr 16;10:796. doi: 10.3389/fimmu.2019.00796. eCollection 2019. Front Immunol. 2019. PMID: 31057538 Free PMC article.
-
Surveying the Metabolic and Dysfunctional Profiles of T Cells and NK Cells in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome.Int J Mol Sci. 2023 Jul 26;24(15):11937. doi: 10.3390/ijms241511937. Int J Mol Sci. 2023. PMID: 37569313 Free PMC article. Review.
-
The search for a blood-based biomarker for Myalgic Encephalomyelitis/ Chronic Fatigue Syndrome (ME/CFS): from biochemistry to electrophysiology.J Transl Med. 2025 Feb 4;23(1):149. doi: 10.1186/s12967-025-06146-6. J Transl Med. 2025. PMID: 39905423 Free PMC article. Review.
Cited by
-
Causal Relationship Between Diet, Lipids, Immune Cells, and Chronic Fatigue Syndrome: A Two-Mediation Mendelian Randomization Study.Food Sci Nutr. 2025 Jun 22;13(6):e70424. doi: 10.1002/fsn3.70424. eCollection 2025 Jun. Food Sci Nutr. 2025. PMID: 40552329 Free PMC article.
-
Biomarkers over Time: From Visual Contrast Sensitivity to Transcriptomics in Differentiating Chronic Inflammatory Response Syndrome and Myalgic Encephalomyelitis/Chronic Fatigue Syndrome.Int J Mol Sci. 2025 Jul 28;26(15):7284. doi: 10.3390/ijms26157284. Int J Mol Sci. 2025. PMID: 40806417 Free PMC article. Review.
References
-
- Choutka J, et al. Unexplained post-acute infection syndromes. Nat Med. 2022;28(5):911–23. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
