

LTP expression mediated by autonomous activity of GluN2B-bound CaMKII
- PMID: 39395168
- PMCID: PMC11563194
- DOI: 10.1016/j.celrep.2024.114866
LTP expression mediated by autonomous activity of GluN2B-bound CaMKII
Abstract
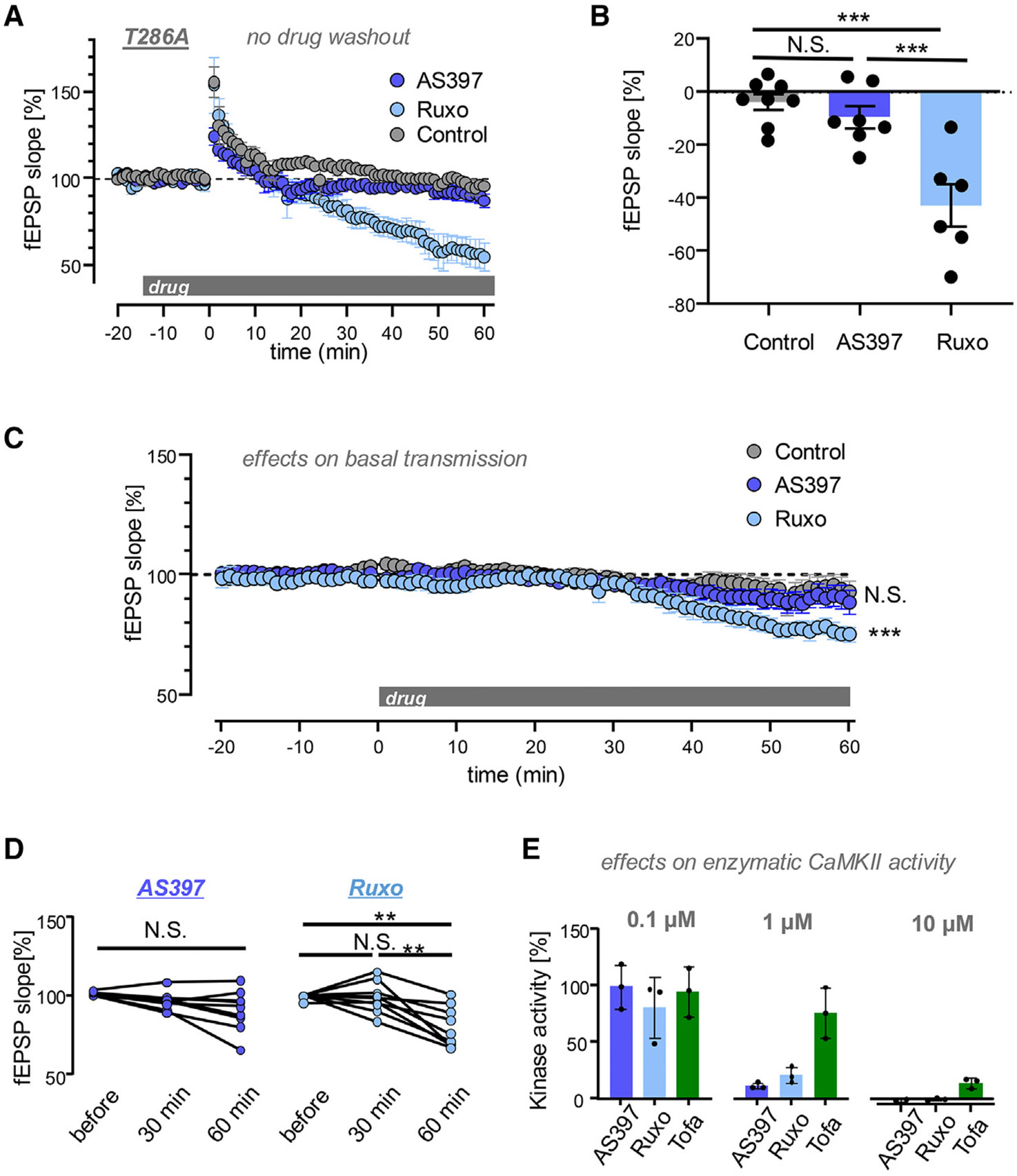
Learning and memory are thought to require the induction and maintenance of long-term potentiation (LTP) of synaptic strength. LTP induction requires the Ca2+/calmodulin-dependent protein kinase II (CaMKII) but for structural rather than enzymatic functions. We show that the relevant structural function is regulated by CaMKII binding to the NMDA-type glutamate receptor subunit GluN2B. This binding directly generates Ca2+-independent autonomous CaMKII activity, and we show that this enzymatic activity is dispensable for LTP induction (within 5 min) but required for a subsequent LTP phase (within 15 min). This requirement for CaMKII activity provides an objective temporal definition for the intermediary phase of LTP expression. Later LTP maintenance may still require structural functions of GluN2B-bound CaMKII but not the resulting enzymatic CaMKII activity or their co-condensation. Thus, autonomous CaMKII activity mediates post-induction LTP but (1) via GluN2B binding, not T286 autophosphorylation, and (2) during the intermediary expression phase rather than for long-term maintenance.
Keywords: CP: Molecular biology; CP: Neuroscience; CaMKII; GluN2B; LTP expression; LTP induction; LTP maintenance; NMDAR; binding; condensation.
Copyright © 2024 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The Regents of the University of Colorado have filed a patent application, with N.L.R., M.E.L., J.E.T., S.J.C., and K.U.B. in the list of inventors, that includes findings reported in this manuscript. K.U.B. is co-founder and board member of Neurexis Therapeutics, a company that seeks to develop a CaMKII inhibitor into a therapeutic drug for cerebral ischemia.
Figures




References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
