

Differential transcriptomic host responses in the early phase of viral and bacterial infections in human lung tissue explants ex vivo
- PMID: 39395995
- PMCID: PMC11471021
- DOI: 10.1186/s12931-024-02988-8
Differential transcriptomic host responses in the early phase of viral and bacterial infections in human lung tissue explants ex vivo
Abstract
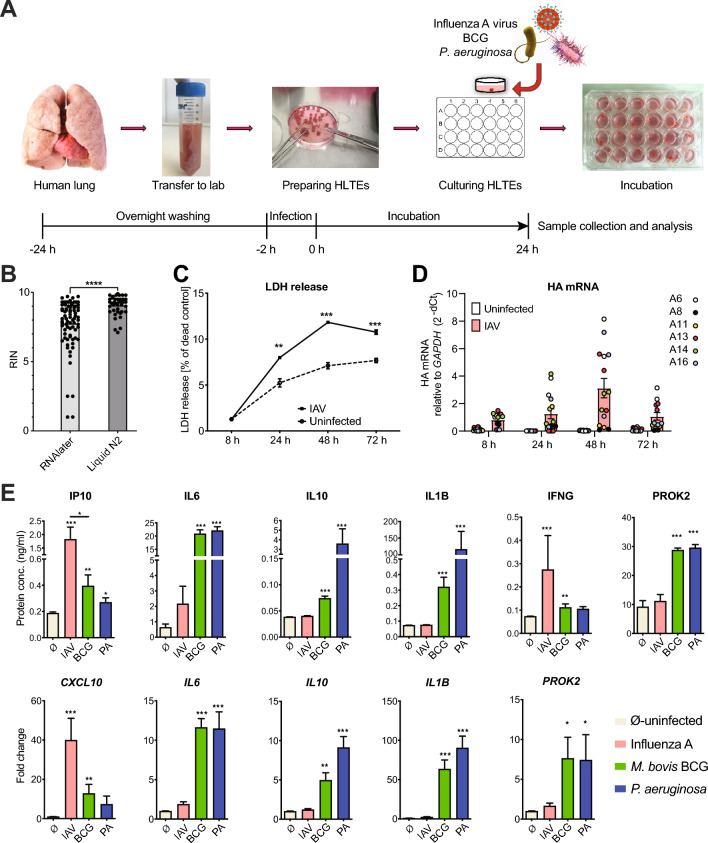
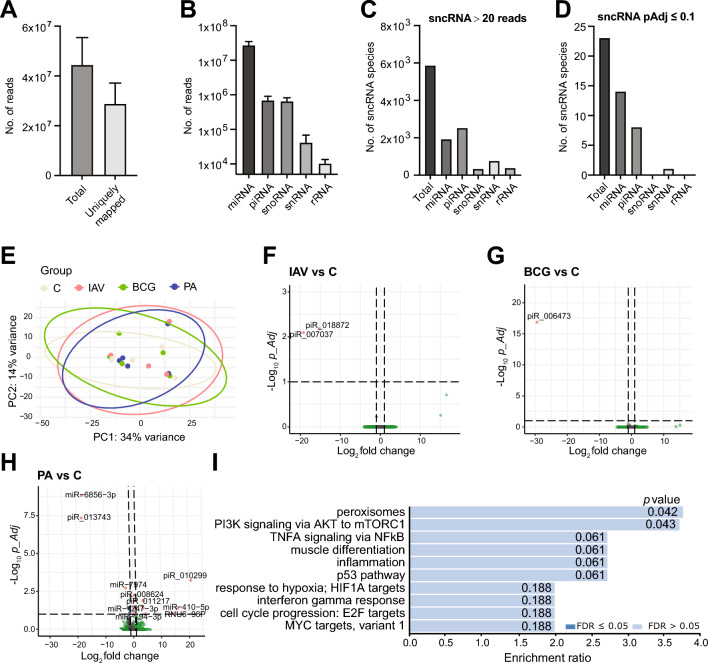
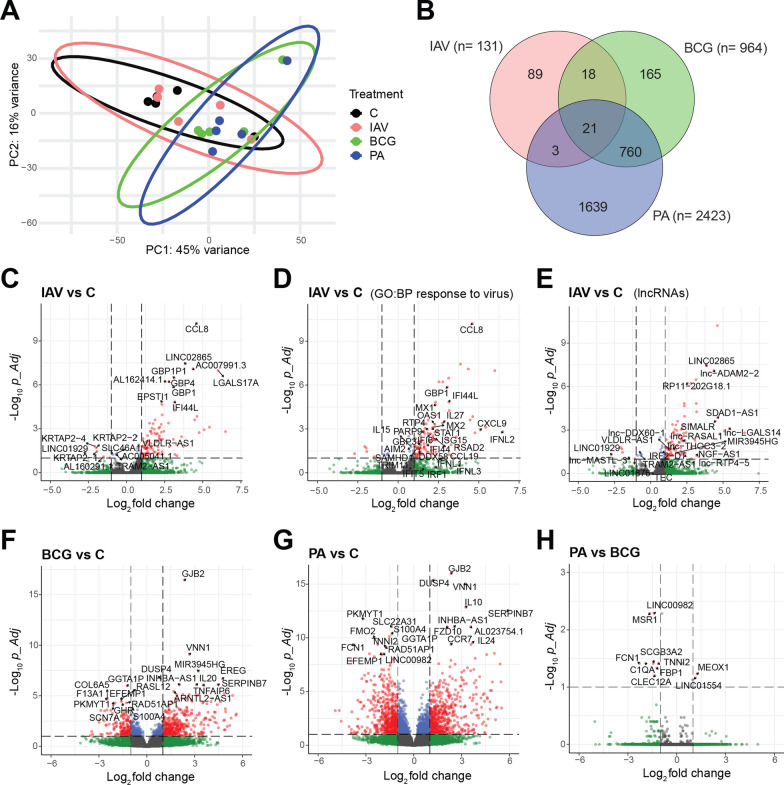
Background: The first 24 h of infection represent a critical time window in interactions between pathogens and host tissue. However, it is not possible to study such early events in human lung during natural infection due to lack of clinical access to tissue this early in infection. We, therefore, applied RNA sequencing to ex vivo cultured human lung tissue explants (HLTE) from patients with emphysema to study global changes in small noncoding RNA, mRNA, and long noncoding RNA (lncRNA, lincRNA) populations during the first 24 h of infection with influenza A virus (IAV), Mycobacterium bovis Bacille Calmette-Guerin (BCG), and Pseudomonas aeruginosa.
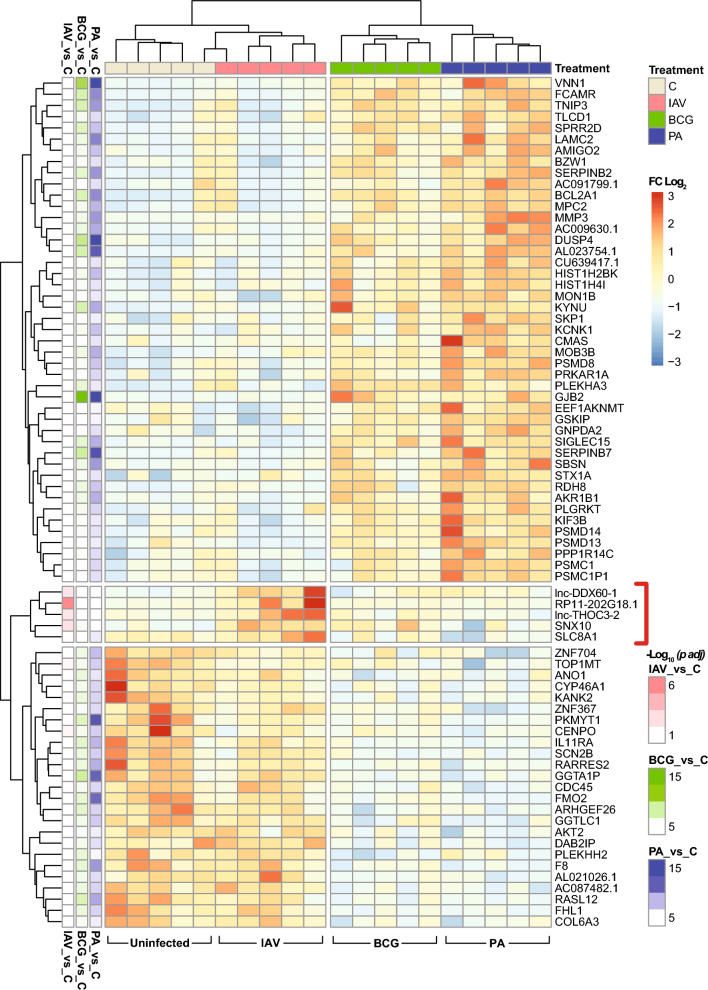
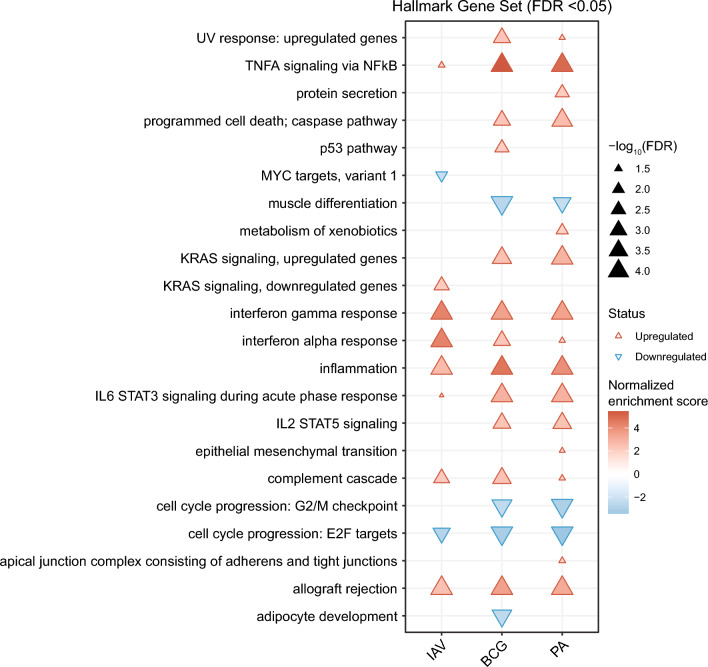
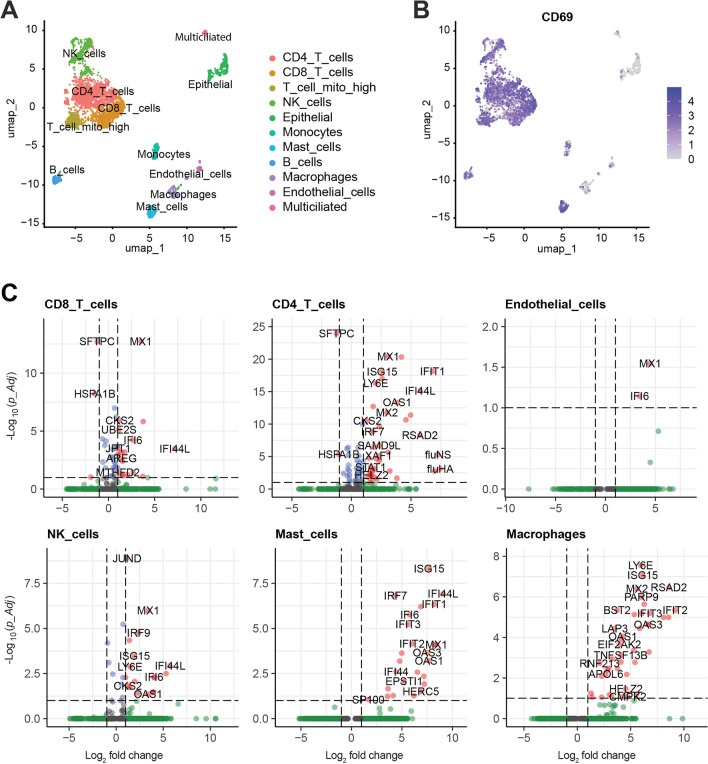
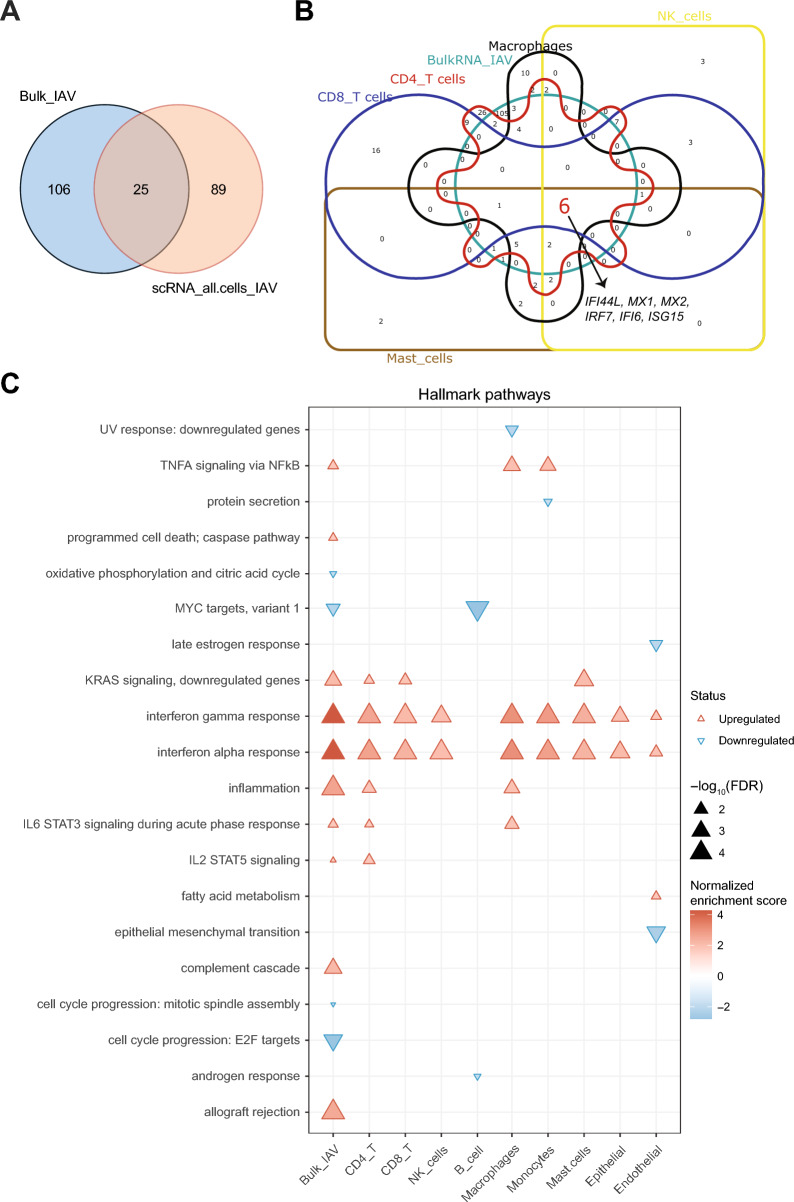
Results: Pseudomonas aeruginosa caused the strongest expression changes and was the only pathogen that notably affected expression of microRNA and PIWI-associated RNA. The major classes of long RNAs (> 100 nt) were represented similarly among the RNAs that were differentially expressed upon infection with the three pathogens (mRNA 77-82%; lncRNA 15-17%; pseudogenes 4-5%), but lnc-DDX60-1, RP11-202G18.1, and lnc-THOC3-2 were part of an RNA signature (additionally containing SNX10 and SLC8A1) specifically associated with IAV infection. IAV infection induced brisk interferon responses, CCL8 being the most strongly upregulated mRNA. Single-cell RNA sequencing identified airway epithelial cells and macrophages as the predominant IAV host cells, but inflammatory responses were also detected in cell types expressing few or no IAV transcripts. Combined analysis of bulk and single-cell RNAseq data identified a set of 6 mRNAs (IFI6, IFI44L, IRF7, ISG15, MX1, MX2) as the core transcriptomic response to IAV infection. The two bacterial pathogens induced qualitatively very similar changes in mRNA expression and predicted signaling pathways, but the magnitude of change was greater in P. aeruginosa infection. Upregulation of GJB2, VNN1, DUSP4, SerpinB7, and IL10, and downregulation of PKMYT1, S100A4, GGTA1P, and SLC22A31 were most strongly associated with bacterial infection.
Conclusions: Human lung tissue mounted substantially different transcriptomic responses to infection by IAV than by BCG and P. aeruginosa, whereas responses to these two divergent bacterial pathogens were surprisingly similar. This HLTE model should prove useful for RNA-directed pathogenesis research and tissue biomarker discovery during the early phase of infections, both at the tissue and single-cell level.
Keywords: Biomarker; Chronic obstructive pulmonary disease; Emphysema; Gene expression; Infection; Long noncoding RNA; Lung tissue; PIWI-associated RNA; Pneumonia; Prokineticin 2; Transcription; miRNA.
© 2024. The Author(s).
Conflict of interest statement
None of the authors have a competing interest relating to conduct of the study or publication of the manuscript.
Figures
References
-
- Sewald K, Danov O. Infection of human precision-cut lung slices with the influenza virus. Methods Mol Biol. 2022;2506:119–34. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
