

[Detection of pathogenic gene mutations in thirteen cases of congenital bilateral absence of vas deferens infertility patients]
- PMID: 39397452
- PMCID: PMC11480543
- DOI: 10.19723/j.issn.1671-167X.2024.05.003
[Detection of pathogenic gene mutations in thirteen cases of congenital bilateral absence of vas deferens infertility patients]
Abstract
Objective: To detect the cystic fibrosis transmembrane transduction regulator (CFTR) gene mutations and congenital bilateral absence of vas deferens (CBAVD) susceptibility gene mutations in patients with CBAVD, and to explore their association with the risk of CBAVD.
Methods: Whole-exome sequencing and Sanger sequencing validation were conducted on the pathogenic genes CFTR, adhesion G protein-coupled receptor G2 (ADGRG2), sodium channel epithelial 1 subunit beta (SCNN1B), carbonic anhydrase 12 (CA12), and solute carrier family 9 member A3 (SLC9A3) in thirteen cases of isolated CBAVD patients. The polymorphic loci, intron and flanking sequences of CFTR gene were amplified by polymerase chain reaction (PCR) followed by Sanger sequencing. Bioinformatics methods were employed for conservative analysis and deleterious prediction of novel susceptibility gene mutations in CBAVD. Genetic analysis was performed on the pedigree of one out of thirteen patients with CBAVD to evaluate the risk of inheritance in offspring.
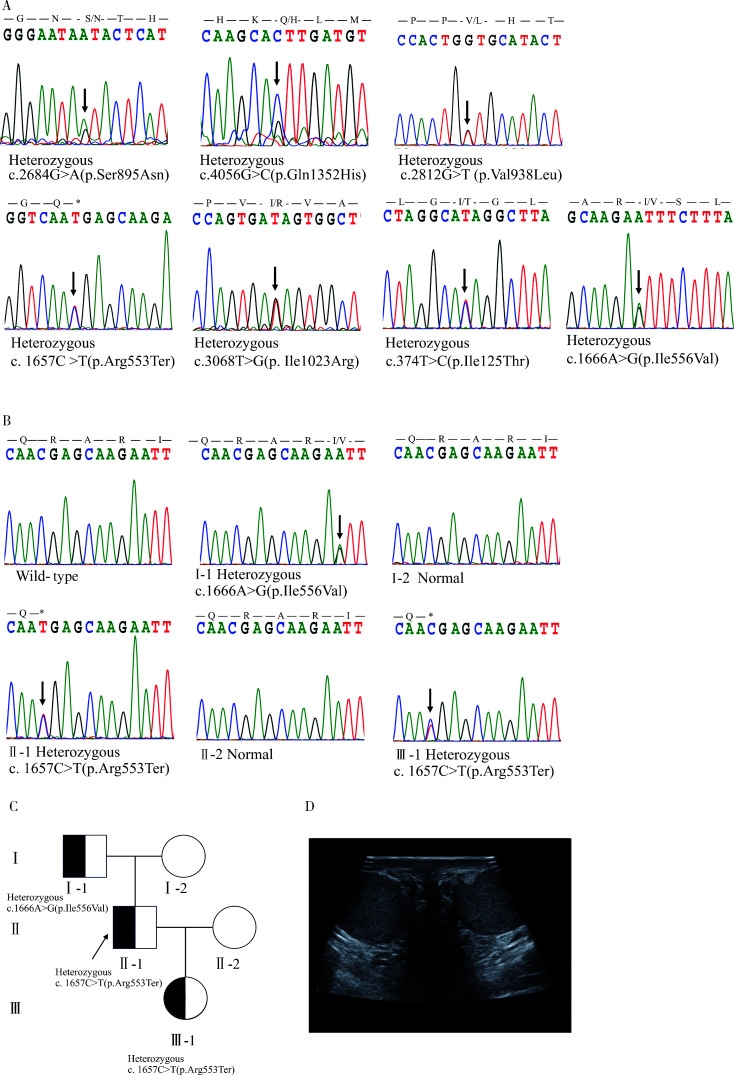
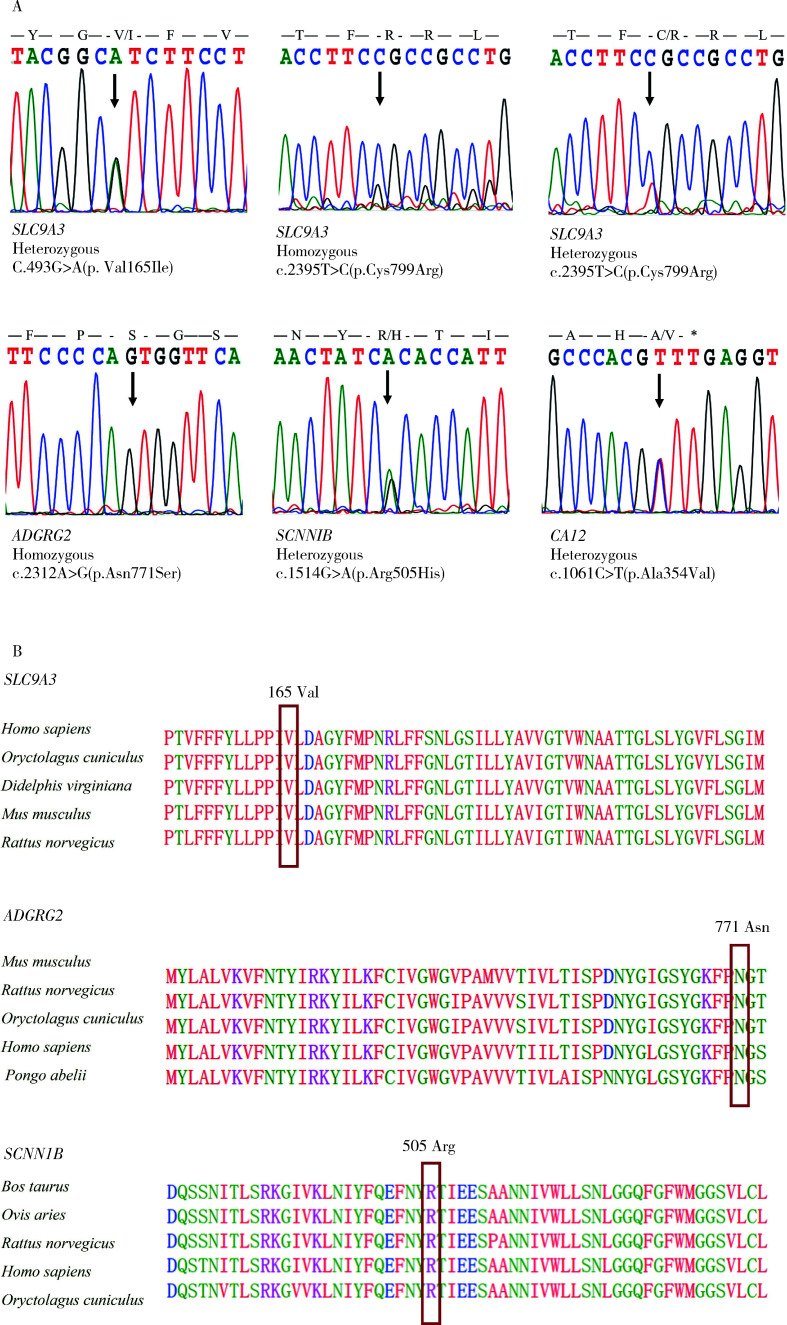
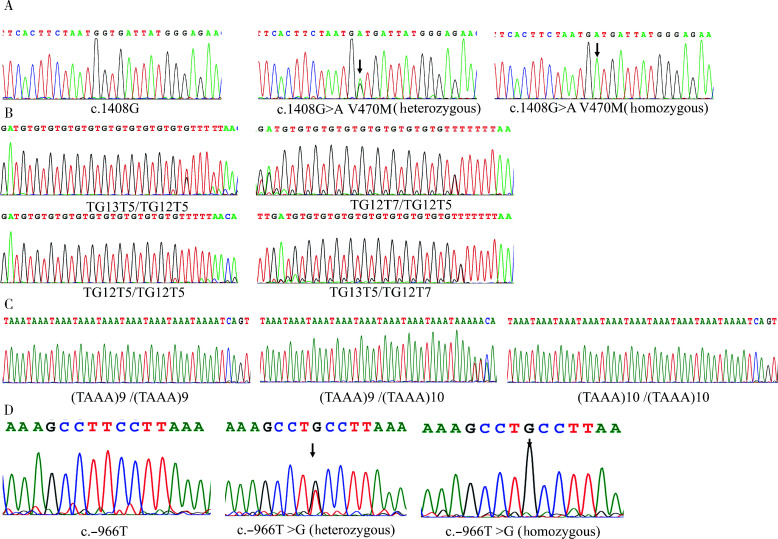
Results: Exome sequencing revealed CFTR gene exon mutations in only six of the thirteen CBAVD patients, with six missense mutations c.2684G>A(p.Ser895Asn), c.4056G>C(p.Gln1352His), c.2812G>(p.Val938Leu), c.3068T>G(p.Ile1023Arg), c.374T>C(p.Ile125Thr), c.1666A>G(p.Ile556Val)), and one nonsense mutation (c.1657C>T(p.Arg553Ter). Among these six patients, two also had the CFTR homozygous p.V470 site, additionally, mutations in CFTR gene exon regions were not detected in the remaining seven patients. Within the thirteen CBAVD patients, three carried the homozygous p.V470 polymorphic site, four carried the 5T allele, two carried the TG13 allele, and ten carried the c.-966T>G site. Four CBAVD patients simultaneously carried 2-3 of the aforementioned CFTR gene mutation sites. Susceptibility gene mutations in CBAVD among the thirteen patients included one ADGRG2 missense mutation c.2312A>G(p.Asn771Ser), two SLC9A3 missense mutations c.2395T>C(p.Cys799Arg), c.493G>A(p.Val165Ile), one SCNN1B missense mutation c.1514G>A(p.Arg505His), and one CA12 missense mutation c.1061C>T (p.Ala354Val). Notably, the SLC9A3 gene c.493G>A (p.Val165Ile) mutation site was first identified in CBAVD patients. The five mutations exhibited an extremely low population mutation frequency in the gnomAD database, classifying them as rare mutations. Predictions from Mutation Taster and Polyphen-2 software indicated that the harmfulness level of the SLC9A3 gene c.493G>A (p.Val165Ile) site and the SCNN1B gene c.1514G>A (p.Arg505His) site were disease causing and probably damaging. The genetic analysis of one pedigree revealed that the c.1657C>T (p.Arg553Ter) mutation in the proband was a de novo mutation, as neither the proband's father nor mother carried this mutation. The proband and his spouse conceived a daughter through assisted reproductive technology, and the daughter inherited the proband's pathogenic mutation c.1657C>T (p.Arg553Ter).
Conclusion: CFTR gene mutations remain the leading cause of CBAVD in Chinese patients; however, the distribution and frequency of mutations differ from data reported in other domestic and international studies, highlighting the need to expand the CFTR mutation spectrum in Chinese CBAVD patients. The susceptibility genes ADGRG2, SLC9A3, SCNN1B, and CA12 may explain some cases of CBAVD without CFTR mutations. Given the lack of specific clinical manifestations in CBAVD patients, it is recommended that clinicians conduct further physical examinations and consider scrotal or transrectal ultrasound before making a defi-nitive diagnosis. It is advisable to employ CFTR gene mutation testing in preconception genetic screening to reduce the risk of CBAVD and cystic fibrosis in offspring.
目的: 检测先天性双侧输精管缺如(congenital bilateral absence of the vas deferens, CBAVD)患者中囊性纤维化跨膜转导调节因子(cystic fibrosis transmembrane conductance regulator, CFTR)基因和CBAVD易感基因的突变情况,探讨它们与CBAVD发病风险的相关性。
方法: 对13例诊断为孤立发生的CBAVD患者的致病基因CFTR及易感基因黏附型G蛋白偶联受体G2(adhesion G protein-coupled receptor G2, ADGRG2)、上皮细胞钠离子通道β亚单位(sodium channel epithelial 1 subunit beta, SCNN1B)和碳酸酐酶12(carbonic anhydrase, CA12)和溶质载体家族9成员3(solute carrier family 9 member A3, SLC9A3)行全外显子测序及Sanger测序验证,针对CFTR基因多态性位点、内含子及侧翼序列行聚合酶链式反应(polymerase chain reaction, PCR)扩增后用Sanger测序,并运用生物信息学方法对CBAVD易感基因新发突变进行保守性分析和有害性预测。对13例CBAVD患者中1例患者的家系进行遗传学分析,评估子代遗传风险。
结果: 外显子测序发现13例CBAVD患者中,只有6例患者检测到CFTR基因外显子突变,有6种错义突变: c.2684G>A(p.Ser895Asn)、c.4056G>C(p.Gln1352His)、c.2812G>T(p.Val938Leu)、c.3068T>G(p.Ile1023Arg)、c.374T>C(p.Ile125Thr)、c.1666A>G(p.Ile556Val), 1种无义突变: c.1657C>T(p.Arg553Ter), 这6例患者中有2例患者同时还存在CFTR的纯合p.V470位点,另外7例患者未检测出CFTR基因外显子区域的突变。13例CBAVD患者中,3例患者携带纯合p.V470的多态性位点,4例患者携带5T等位基因,2例患者携带TG13等位基因,10例患者携带c.-966T>G位点。4例CBAVD患者同时携带以上CFTR基因突变位点中的2~3个位点。13例患者中CBAVD易感基因突变情况: 1种ADGRG2错义突变c.2312A>G(p.Asn771Ser),2种SLC9A3错义突变c.2395T>C(p.Cys799Arg)、c.493G>A(p.Val165Ile), 1种SCNN1B错义突变c.1514G>A(p.Arg505His)和1种CA12错义突变c.1061C>T(p.Ala354Val), 其中,SLC9A3基因的c.493G>A(p.Val165Ile)突变位点是首次在CBAVD患者中被发现,以上5种突变位点在gnomAD数据库中的人群变异频率极低,属于罕见突变,用Mutation Taster和Polyphen-2软件预测显示SLC9A3基因的c.493G>A(p.Val165Ile)位点和SCNN1B基因的c.1514G>A(p.Arg505His)位点的有害性等级为致病突变。1例家系遗传分析发现,先证者的c.1657C>T(p.Arg553Ter)突变为新生突变,先证者父亲、母亲均未携带该突变,先证者及其配偶通过辅助生殖技术孕育1女婴,该女婴遗传了先证者的致病性突变c.1657C>T(p.Arg553Ter)。
结论: CFTR基因突变仍然是中国CBAVD患者的主要致病原因,但突变的分布与频率与国内外其他研究的数据存在一定差异,需要进一步扩充中国CBAVD患者的CFTR突变谱; ADGRG2、SLC9A3、SCNN1B和CA12易感基因可能解释部分无CFTR突变的CBAVD病例; CBAVD患者多无特殊临床表现,建议临床医生确诊前对患者行进一步的体格检查,并结合其阴囊超声或经直肠超声检查; 建议将CFTR基因突变检测应用于辅助生殖前的遗传学筛查,降低子代罹患CBAVD及囊性纤维化的风险。
Keywords: Adhesion G protein-coupled receptor G2; Congenital bilateral absence of the vas deferens; Cystic fibrosis transmembrane conduc-tance regulator; Solute carrier family 9 member A3.
Conflict of interest statement
Figures
Similar articles
-
Genetic mutation analysis of 22 patients with congenital absence of vas deferens: a single-center study†.Biol Reprod. 2022 Jan 13;106(1):108-117. doi: 10.1093/biolre/ioab194. Biol Reprod. 2022. PMID: 34673937
-
Compound heterozygous mutations in CFTR causing CBAVD in Chinese pedigrees.Mol Genet Genomic Med. 2018 Nov;6(6):1097-1103. doi: 10.1002/mgg3.486. Epub 2018 Nov 18. Mol Genet Genomic Med. 2018. PMID: 30450785 Free PMC article.
-
Genetic diagnosis and sperm retrieval outcomes for Chinese patients with congenital bilateral absence of vas deferens.Andrology. 2020 Sep;8(5):1064-1069. doi: 10.1111/andr.12769. Epub 2020 Feb 25. Andrology. 2020. PMID: 32020786
-
The role of SLC9A3 in Taiwanese patients with congenital bilateral absence of vas deferens (CBAVD).J Formos Med Assoc. 2019 Dec;118(12):1576-1583. doi: 10.1016/j.jfma.2019.01.019. Epub 2019 Feb 21. J Formos Med Assoc. 2019. PMID: 30797621 Review.
-
[Gene mutations in congenital bilateral absence of the vas deferens: An update].Zhonghua Nan Ke Xue. 2021 May;27(5):450-455. Zhonghua Nan Ke Xue. 2021. PMID: 34914322 Review. Chinese.
References
-
- 王 宇刚, 马 淑霞, 姚 晓飞, et al. 男性不育伴精液量少的临床特点分析. 生殖医学杂志. 2023;32(7):997–1002. doi: 10.3969/j.issn.1004-3845.2023.07.005. - DOI
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous