

The Spectrum of clinical manifestations in newborns with the COQ4 mutation: case series and literature review
- PMID: 39398416
- PMCID: PMC11466766
- DOI: 10.3389/fped.2024.1410133
The Spectrum of clinical manifestations in newborns with the COQ4 mutation: case series and literature review
Abstract
Background: Coenzyme Q10 (CoQ10) plays an important role in the electron transport chain within the human mitochondrial respiratory chain. The manifestations of this deficiency exhibit a diverse range. This study investigates the clinical manifestations of primary coenzyme Q10 deficiency in neonates with the COQ4 mutation to improve the diagnosis of the disease and the prognosis through targeted treatment.
Methods: We report 4 patients with primary coenzyme Q10 deficiency by COQ4 variants in neonates. A comprehensive literature search and review for original articles and case reports with COQ4 mutation published from January 1989 to November 2023 was performed through Pubmed. We review clinical manifestations, diagnostic approaches, and treatment monitoring in these and 20 previously reported patients.
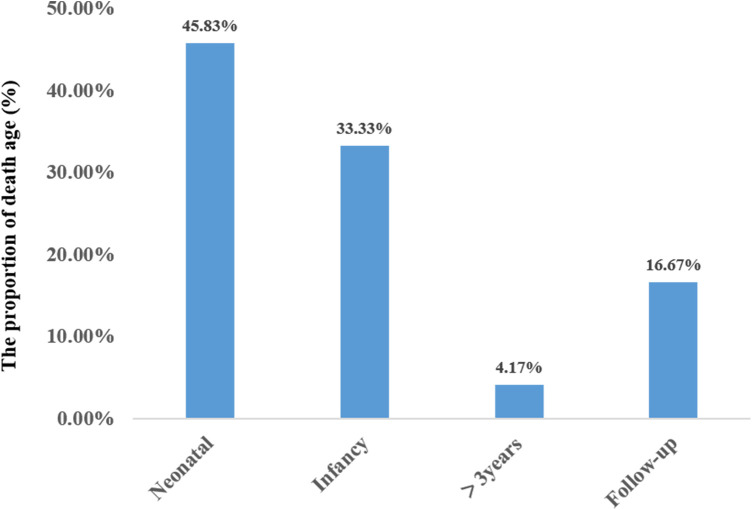
Results: Within the cohort of four cases examined, three females and one male were identified from two distinct families. Specifically, case 1 and 2 consisted of monoamniotic twins. Cases 3 and 4 were siblings. A comprehensive review of 20 cases involving neonatal-onset COQ4 mutation was conducted. Half of the cases are Chinese. There was no statistically significant difference in the mortality between Chinese (9/12, 75%) and other regions (11/12, 91.7%) (P = 0.27). The survival time for the 24 cases was 60.0 ± 98.0 days (95% confidence interval CI: 0-252.0 days). The incidence of prenatal abnormalities in preterm infants was significantly higher than that in full-term infants (66.7% vs. 16.7%, P = 0.02). Hyperlactatemia was one of the most common manifestations, accounting for 75% of cases (18/24). Twenty of the 24 cases were diagnosed by whole exome sequencing. Only 9 patients received exogenous coenzyme Q10 treatment, and all the 4 surviving patients received coenzyme Q10 supplementation.
Conclusion: The prognosis of COQ4 mutation in the neonatal period indicates a low survival rate and an poor prognosis. This may be due to the incomplete understanding of the mechanism of how COQ4 gene defects lead to coenzyme Q10 deficiency and why CoQ10 supplementation does not respond well to treatment. To improve the diagnostic rate, in addition to genetic testing, mitochondrial functional verification should be prioritized in southern China, where the incidence is relatively high. It will facilitate more in-depth mechanistic studies.
Keywords: COQ4; CoQ10; mitochondrial disorder; newborn; primary coenzyme Q10 deficiency.
© 2024 Pan, Zhou, Sun, Chen, Han and Zhou.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures





Similar articles
-
Primary Coenzyme Q10 Deficiency-7 and Pathogenic COQ4 Variants: Clinical Presentation, Biochemical Analyses, and Treatment.Front Genet. 2022 Jan 26;12:776807. doi: 10.3389/fgene.2021.776807. eCollection 2021. Front Genet. 2022. PMID: 35154243 Free PMC article. Review.
-
Two Turkish patients with Primary Coenzyme Q10 Deficiency-7: case report and literature review.J Pediatr Endocrinol Metab. 2024 Feb 2;37(3):260-270. doi: 10.1515/jpem-2023-0490. Print 2024 Mar 25. J Pediatr Endocrinol Metab. 2024. PMID: 38353291 Review.
-
Epilepsy and Coenzyme Q10 deficiency with COQ4 variants.Epilepsy Behav. 2023 Dec;149:109498. doi: 10.1016/j.yebeh.2023.109498. Epub 2023 Nov 9. Epilepsy Behav. 2023. PMID: 37948995
-
[Primary coenzyme Q10 deficiency-7: a case report and literature review].Zhonghua Er Ke Za Zhi. 2020 Nov 2;58(11):928-932. doi: 10.3760/cma.j.cn112140-20200610-00601. Zhonghua Er Ke Za Zhi. 2020. PMID: 33120466 Review. Chinese.
-
COQ4 mutations cause a broad spectrum of mitochondrial disorders associated with CoQ10 deficiency.Am J Hum Genet. 2015 Feb 5;96(2):309-17. doi: 10.1016/j.ajhg.2014.12.023. Am J Hum Genet. 2015. PMID: 25658047 Free PMC article.
References
LinkOut - more resources
Full Text Sources