

Spinal Muscular Atrophy Update in Best Practices: Recommendations for Treatment Considerations
- PMID: 39399564
- PMCID: PMC11464225
- DOI: 10.1212/CPJ.0000000000200374
Spinal Muscular Atrophy Update in Best Practices: Recommendations for Treatment Considerations
Abstract
Background and objectives: Spinal muscular atrophy (SMA) is an autosomal recessive disorder caused by biallelic variants of the Survival Motor Neuron 1 gene (SMN1) that affects approximately 1 in 15,000 live births. Availability of 3 SMN-enhancing treatments for SMA has led to urgency to review how clinicians and patients use these treatments for SMA, while additional research and real-world data and experience are being collected. This work describes important factors to assist with decision-making for SMN-enhancing treatments.
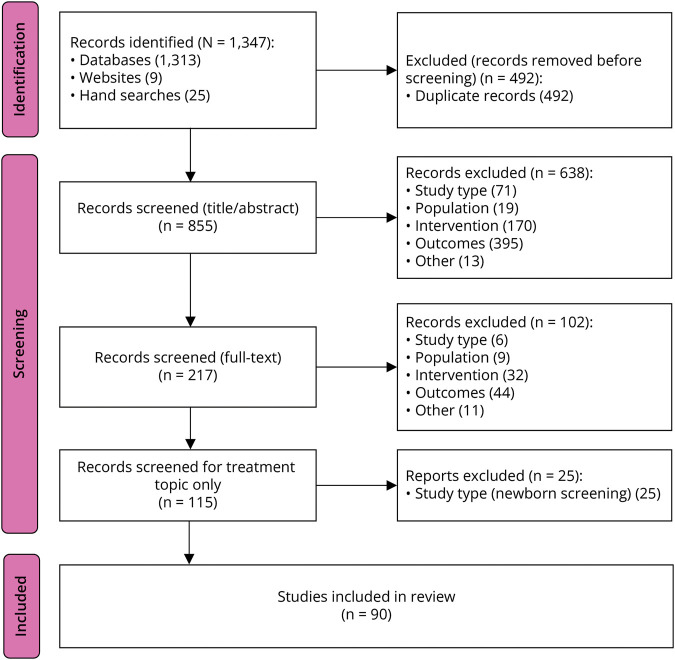
Methods: A systematic literature review was conducted on SMN-enhancing treatments for SMA and related studies. A working group of American and European health care providers with expertise in SMA care identified barriers and developed recommendations through a modified Delphi technique with serial surveys and feedback through virtual meetings to fill gaps for information where evidence is limited. A community working group of an individual living with SMA and caregivers provided insight and perspective on SMA treatments and support through a virtual meeting to guide recommendations.
Results: The health care provider working group and the community working group agreed that when determining whether to start, change, add, or discontinue a treatment, essential considerations include patient and family/caregiver perspective, and treatment safety and side effects. When initiating treatment for patients newly diagnosed with SMA, important patient characteristics are age and Survival Motor Neuron 2 gene (SMN2) copy number. Furthermore, when initiating, changing, or adding treatment, current clinical status and comorbidities drive decision-making. When considering a medication or treatment plan change, unless there is an urgent indication, a treatment and associated patient outcomes should be monitored for a minimum of 6-12 months. When determining a treatment plan with an adolescent or adult with SMA, consider factors such as quality of life, burden vs benefit of treatment, and reproductive issues. Access to care coordination and interdisciplinary/multidisciplinary care are essential to treatment success.
Discussion: Sharing information about current knowledge of treatments and shared decision-making between health care providers and patients living with SMA and caregivers are essential to overcoming barriers to providing SMN-enhancing treatments.
Copyright © 2024 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology.
Conflict of interest statement
M.K. Schroth is an employee of Cure SMA; J. Deans is an employee of Cure SMA; D.X. Bharucha-Goebel has received research support from Genentech/Roche; W.B. Burnette reports no disclosures; B.T. Darras has served as an ad hoc scientific advisory board member for AveXis/Novartis Gene Therapies, Biogen, and Roche/Genentech; Steering Committee Chair/Member for Roche FIREFISH and MANATEE studies, and has received research support from Biogen, Novartis Gene Therapies (AveXis), and Roche/Genentech; B.H. Elsheikh received research support from Biogen, Genentech, NMD Pharma, and served as a consultant for Biogen and Genentech; M.V. Felker served as an advisor for PHAR (Partnership for Health Analytic Research), which was hired by Genentech and also received honoraria serving as a consultant for Genentech; A. Klein received honoraria for serving on advisory boards and/or symposia for Biogen, Novartis, and Roche, has received research support from Biogen, Novartis, and Roche, and serves as clinical lead of the Swiss-Reg NMD; J. Krueger received research support and served as a primary investigator for clinical trials sponsored by Novartis, Genetech/Roche, and Scholar Rock; C.M. Proud has received research support and served as a site primary investigator for clinical trials sponsored by Biogen, Novartis Gene Therapies, and Scholar Rock, has received honoraria for serving on advisory board or Speaker's Bureaus for Biogen, Novartis Gene Therapies, and Roche; A. Veerapandiyan has received honoraria for serving on ad hoc advisory boards for Biogen and for Novartis, and receives research support from the Muscular Dystrophy Association and from Novartis; R.J. Graham reports no disclosures. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/cp.
Figures
References
-
- Feldkotter M, Schwarzer V, Wirth R, Wienker TF, Wirth B. Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet. 2002;70(2):358-368. doi: 10.1086/338627 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources