

Mapping extrachromosomal DNA amplifications during cancer progression
- PMID: 39402156
- PMCID: PMC11549044
- DOI: 10.1038/s41588-024-01949-7
Mapping extrachromosomal DNA amplifications during cancer progression
Abstract
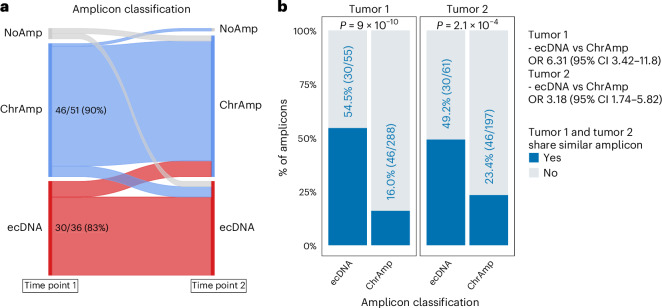
To understand the role of extrachromosomal DNA (ecDNA) amplifications in cancer progression, we detected and classified focal amplifications in 8,060 newly diagnosed primary cancers, untreated metastases and heavily pretreated tumors. The ecDNAs were detected at significantly higher frequency in untreated metastatic and pretreated tumors compared to newly diagnosed cancers. Tumors from chemotherapy-pretreated patients showed significantly higher ecDNA frequency compared to untreated cancers. In particular, tubulin inhibition associated with ecDNA increases, suggesting a role for ecDNA in treatment response. In longitudinally matched tumor samples, ecDNAs were more likely to be retained compared to chromosomal amplifications. EcDNAs shared between time points, and ecDNAs in advanced cancers were more likely to harbor localized hypermutation events compared to private ecDNAs and ecDNAs in newly diagnosed tumors. Relatively high variant allele fractions of ecDNA localized hypermutations implicated early ecDNA mutagenesis. Our findings nominate ecDNAs to provide tumors with competitive advantages during cancer progression and metastasis.
© 2024. The Author(s).
Conflict of interest statement
R.G.W.V. is a cofounder of, holds equity in and has received research funds from Boundless Bio. H.K. has received research funds from JW Pharmaceutical. J.L. receives compensation as a part-time consultant for Boundless Bio. V.B. is a cofounder, paid consultant and Scientific Advisory Board member, and has an equity interest in Boundless Bio and Abterra. The terms of this arrangement have been reviewed and approved by the University of California, San Diego, in accordance with its conflict-of-interest policies. All other authors declare no competing interests.
Figures














References
MeSH terms
Grants and funding
- R21 NS114873/NS/NINDS NIH HHS/United States
- OT2 CA278649/CA/NCI NIH HHS/United States
- U24 CA264379/CA/NCI NIH HHS/United States
- P30 CA034196/CA/NCI NIH HHS/United States
- CGCATF-2021/100012/CRUK_/Cancer Research UK/United Kingdom
- CGCATF-2021/100025/Cancer Research UK (CRUK)
- NRF-2019R1A5A2027340/National Research Foundation of Korea (NRF)
- R33 CA236681/CA/NCI NIH HHS/United States
- R01 CA237208/CA/NCI NIH HHS/United States
- OT2 CA278635/CA/NCI NIH HHS/United States
- HI19C1348/Korea Health Industry Development Institute (KHIDI)
- U24CA264379/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- OT2 CA278688/CA/NCI NIH HHS/United States
- CGCATF-2021/100016/Cancer Research UK (CRUK)
- NRF-2022M3C1A3092022/National Research Foundation of Korea (NRF)
- OT2CA278635/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- R01 GM114362/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
