

Integrated multimodal cell atlas of Alzheimer's disease
- PMID: 39402379
- PMCID: PMC11614693
- DOI: 10.1038/s41593-024-01774-5
Integrated multimodal cell atlas of Alzheimer's disease
Abstract
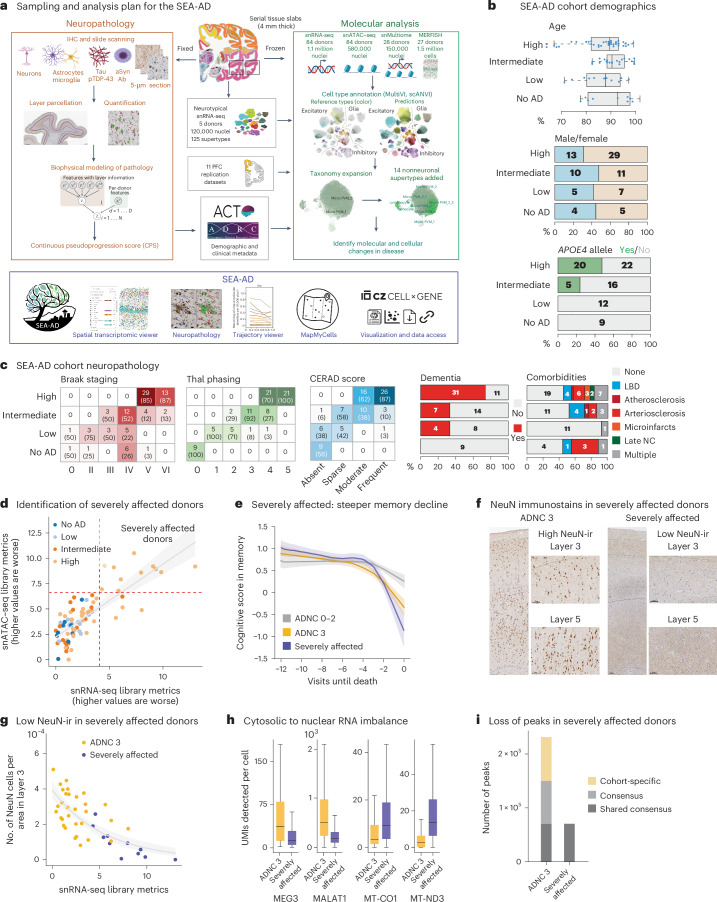
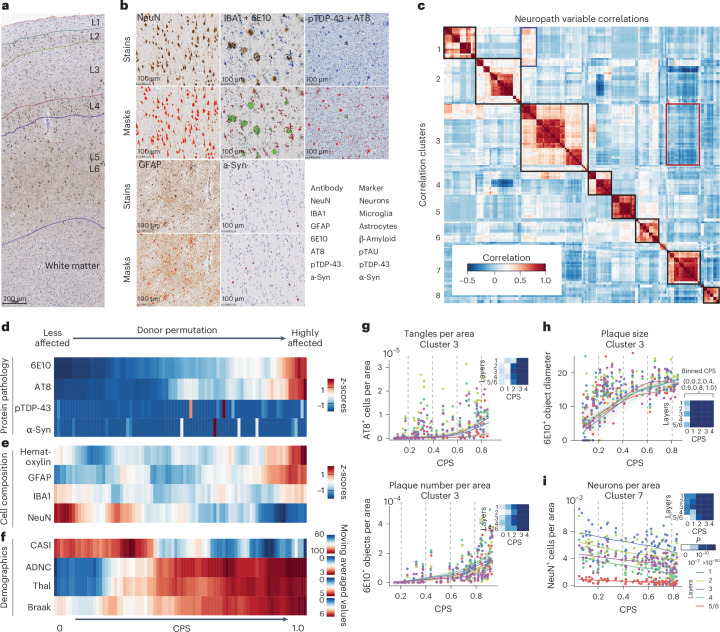
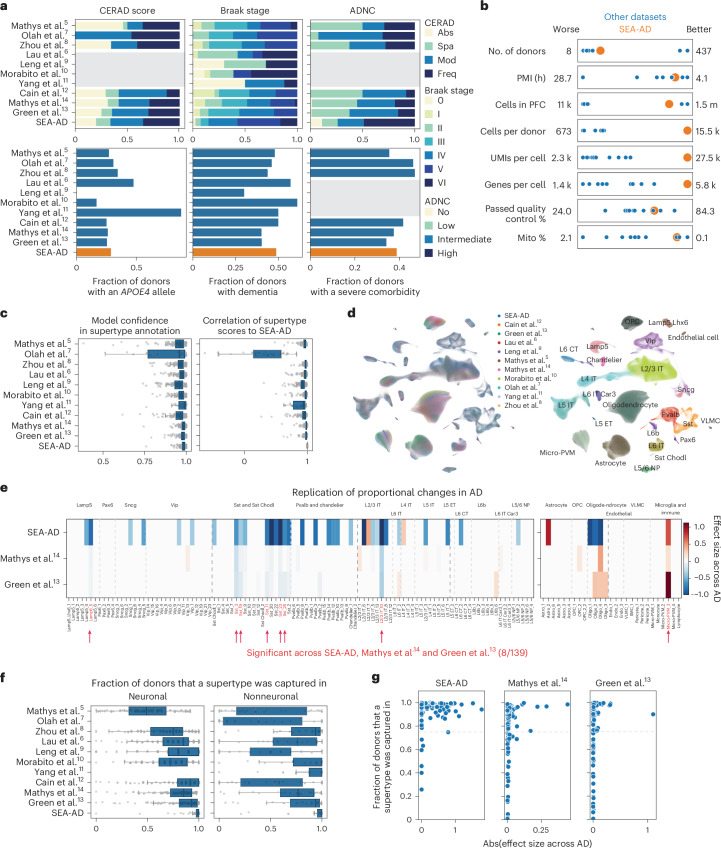
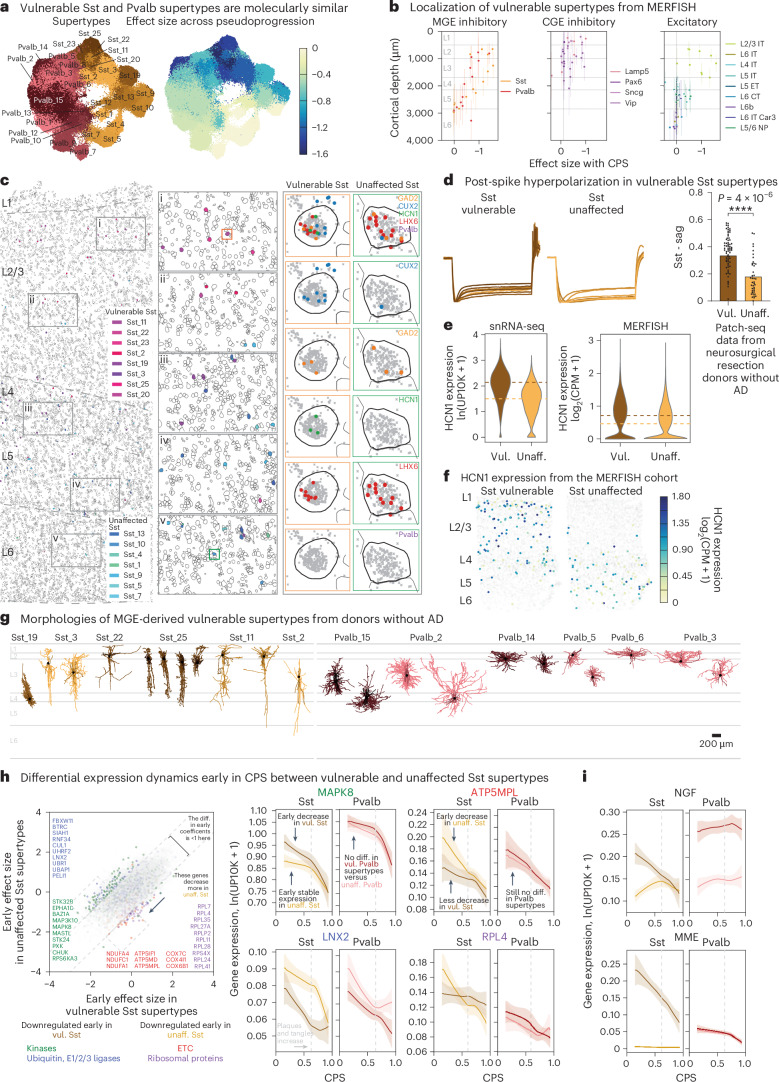
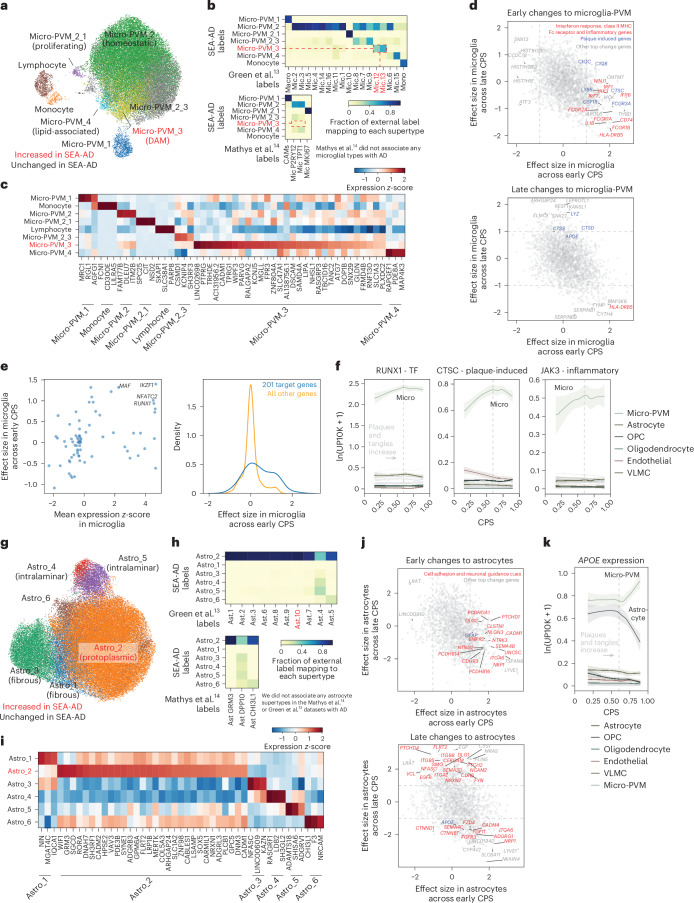
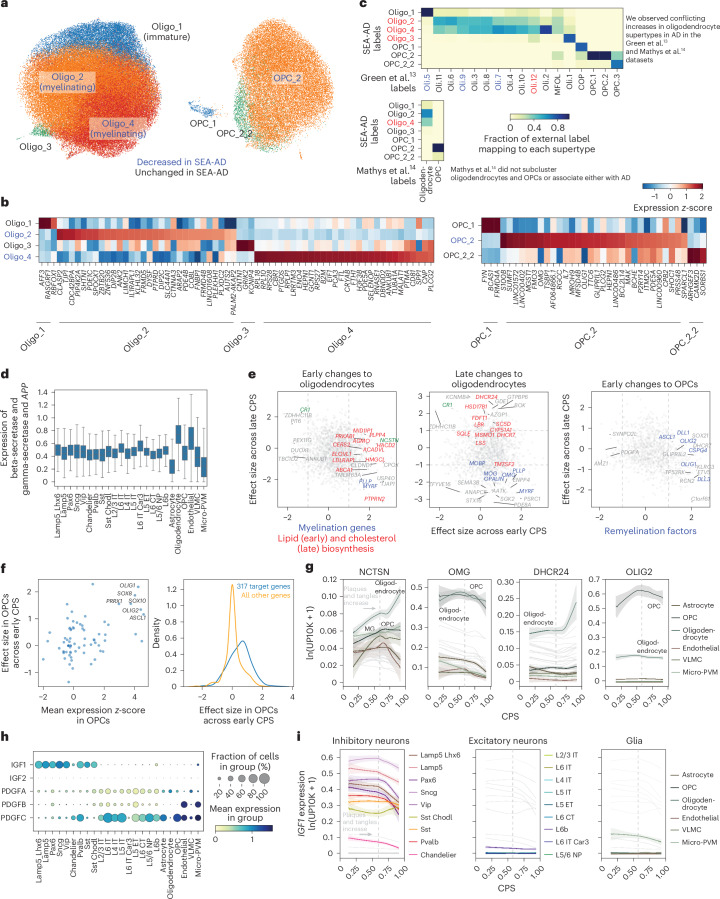
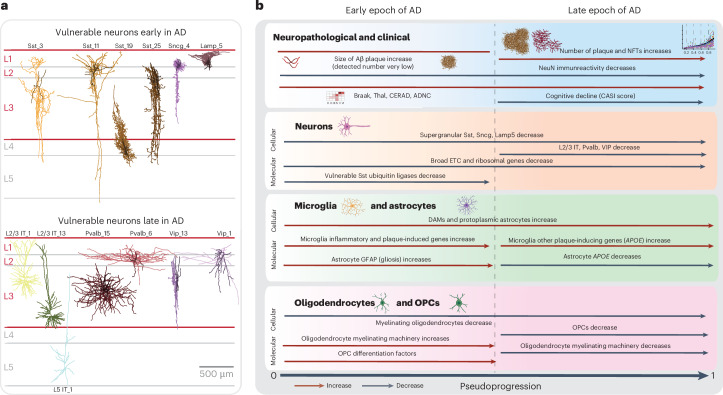
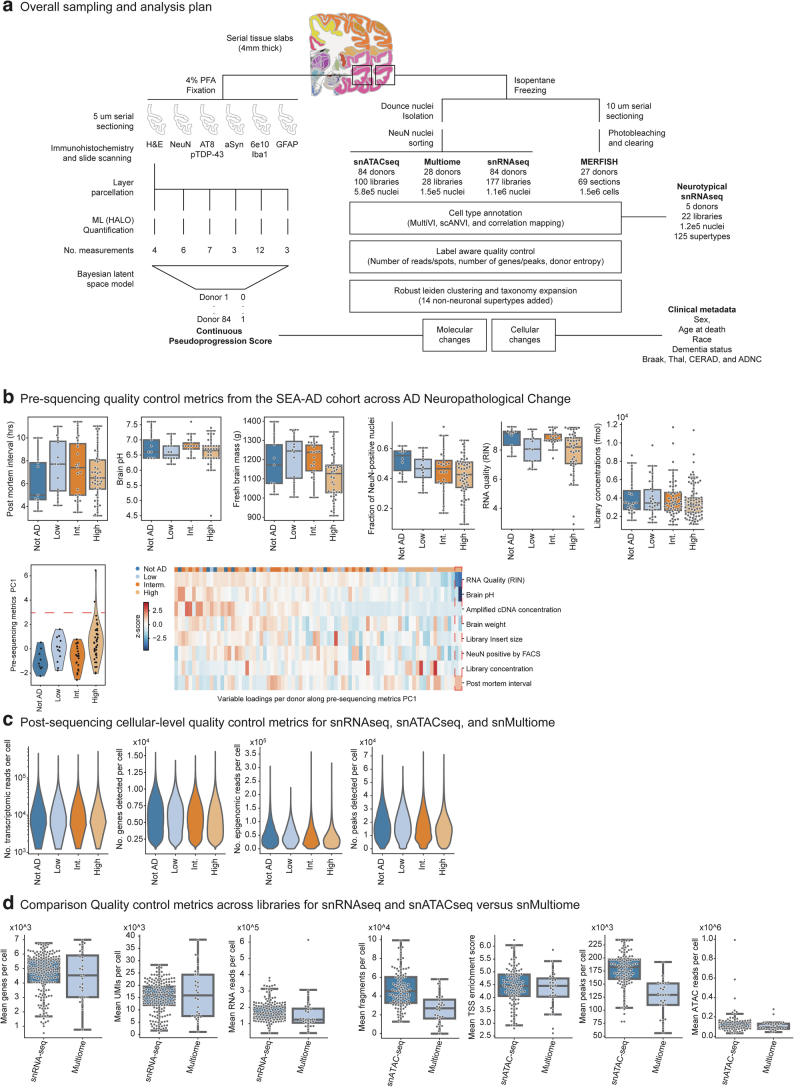
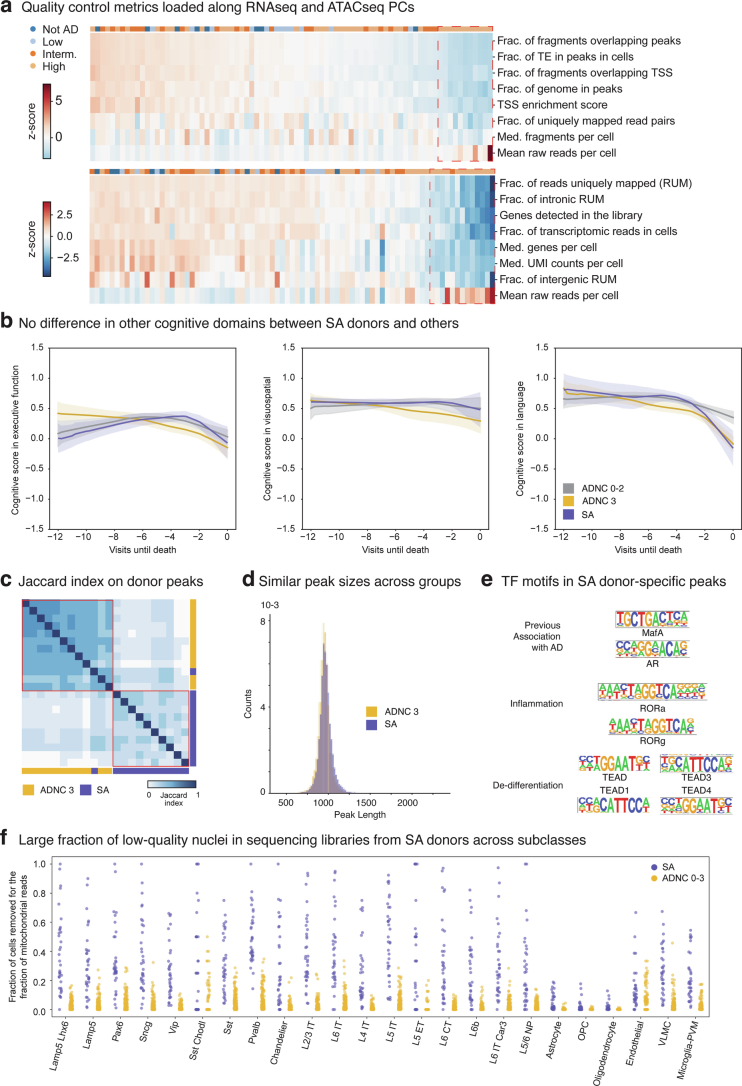
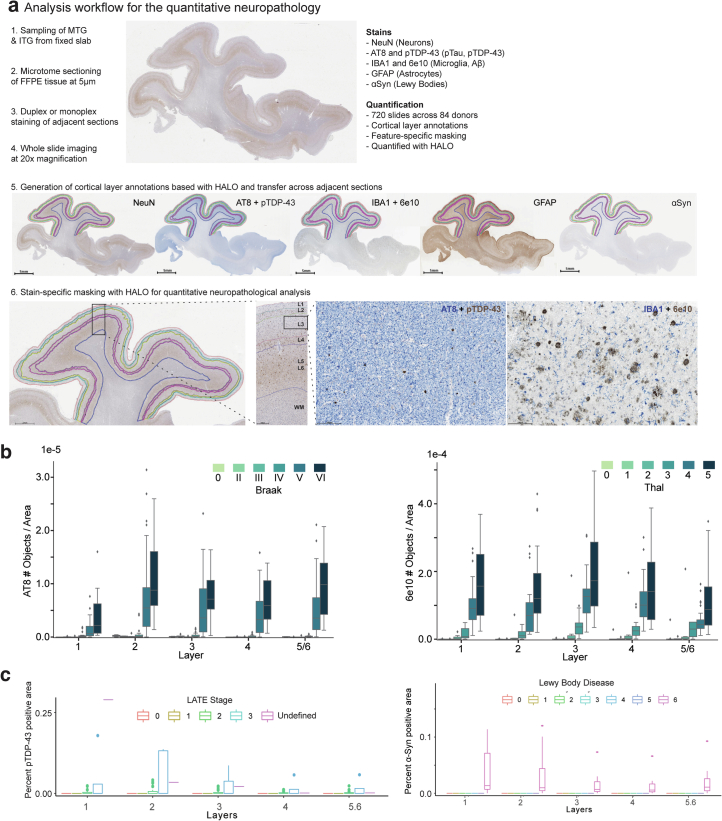
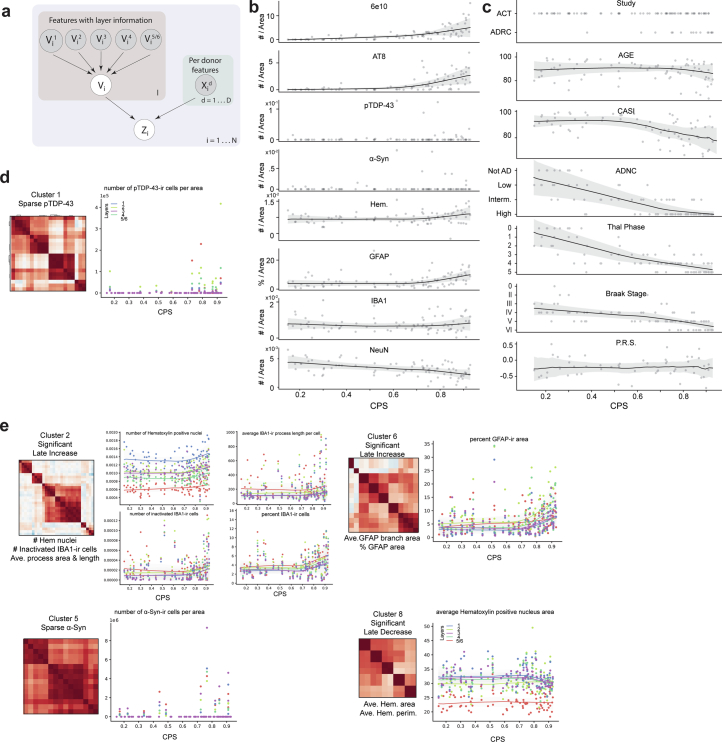
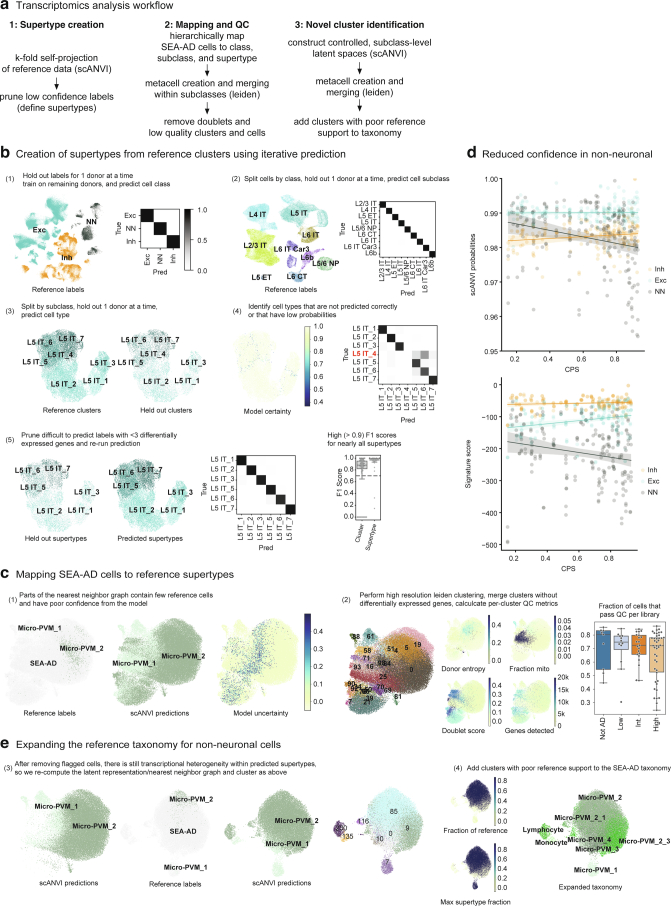
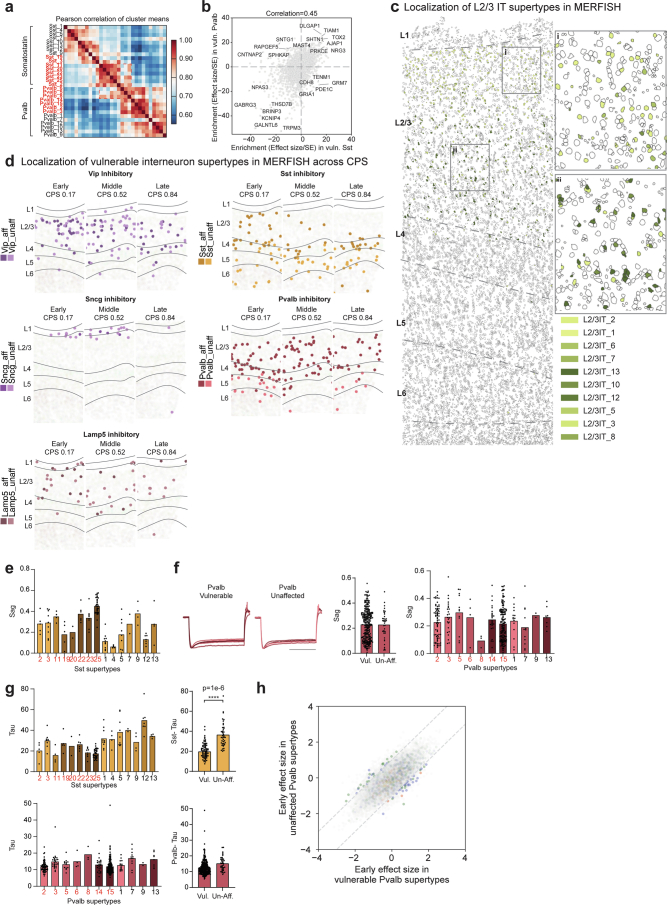
Alzheimer's disease (AD) is the leading cause of dementia in older adults. Although AD progression is characterized by stereotyped accumulation of proteinopathies, the affected cellular populations remain understudied. Here we use multiomics, spatial genomics and reference atlases from the BRAIN Initiative to study middle temporal gyrus cell types in 84 donors with varying AD pathologies. This cohort includes 33 male donors and 51 female donors, with an average age at time of death of 88 years. We used quantitative neuropathology to place donors along a disease pseudoprogression score. Pseudoprogression analysis revealed two disease phases: an early phase with a slow increase in pathology, presence of inflammatory microglia, reactive astrocytes, loss of somatostatin+ inhibitory neurons, and a remyelination response by oligodendrocyte precursor cells; and a later phase with exponential increase in pathology, loss of excitatory neurons and Pvalb+ and Vip+ inhibitory neuron subtypes. These findings were replicated in other major AD studies.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures

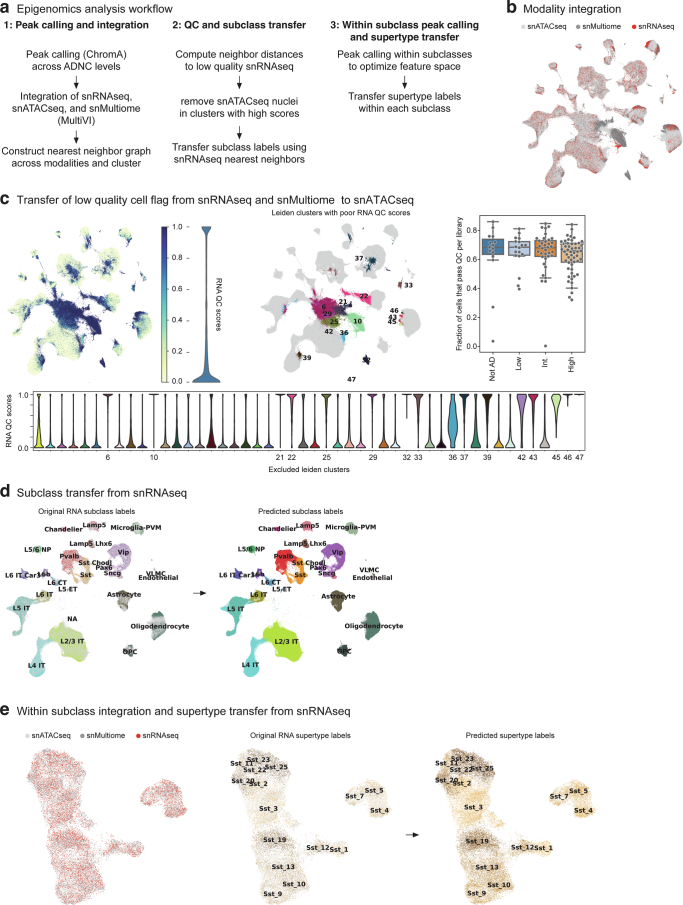
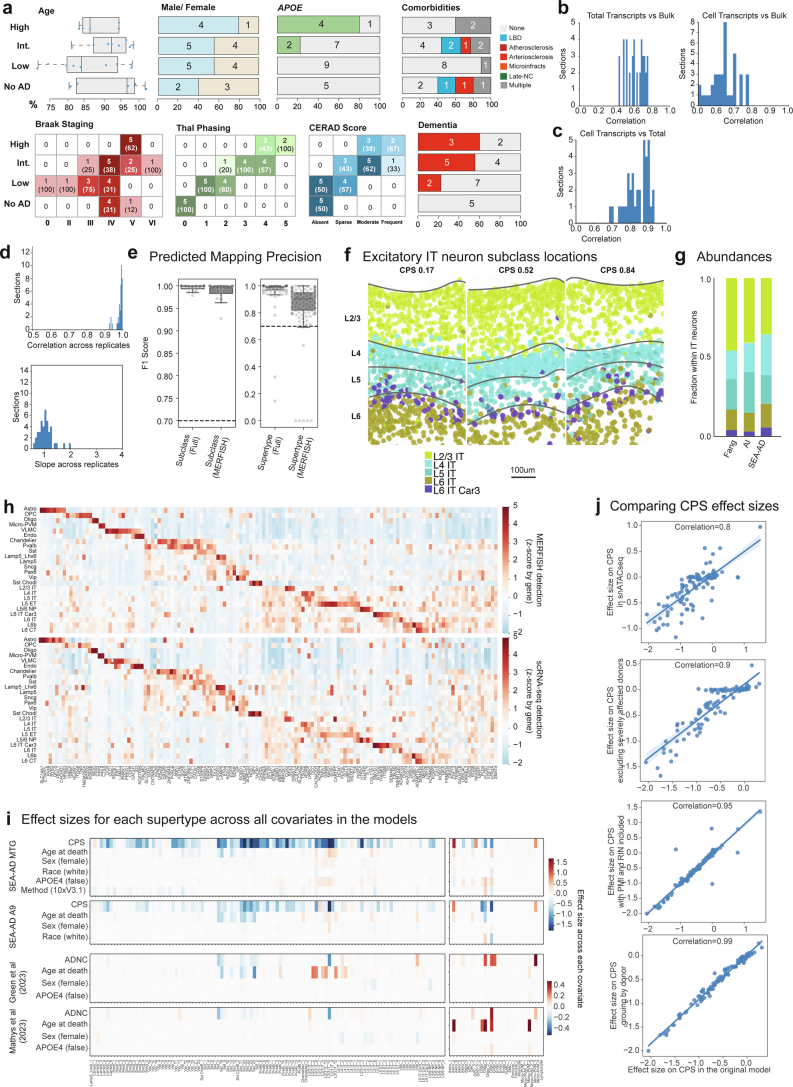
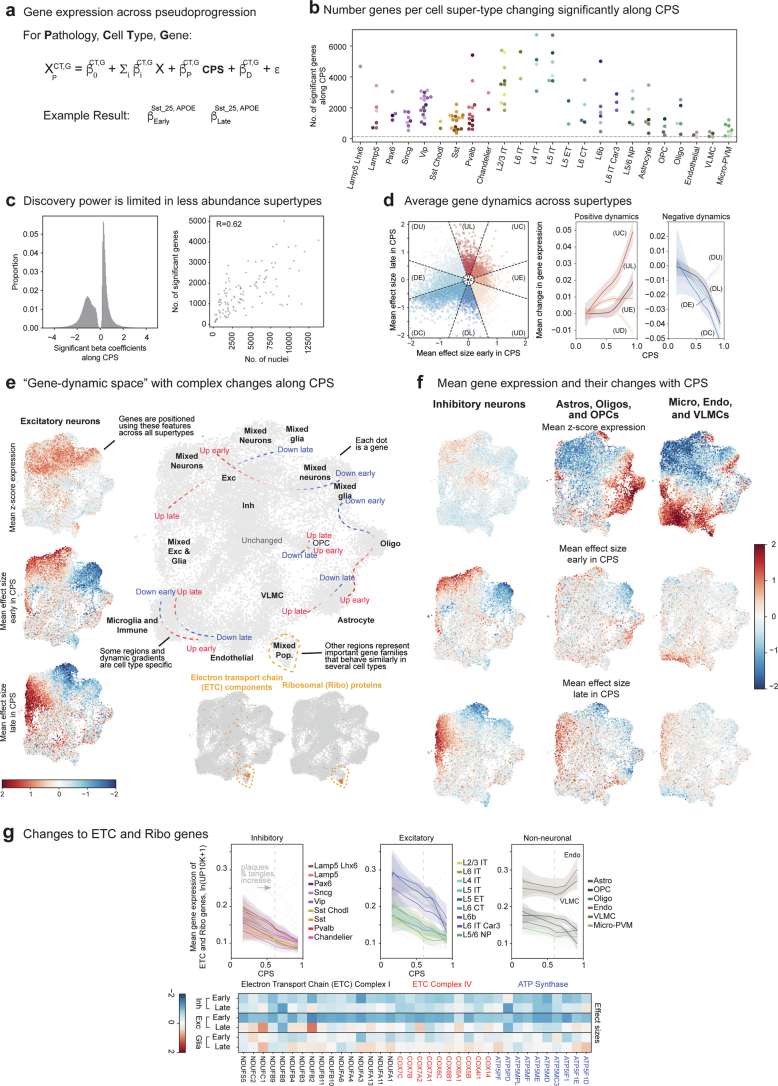
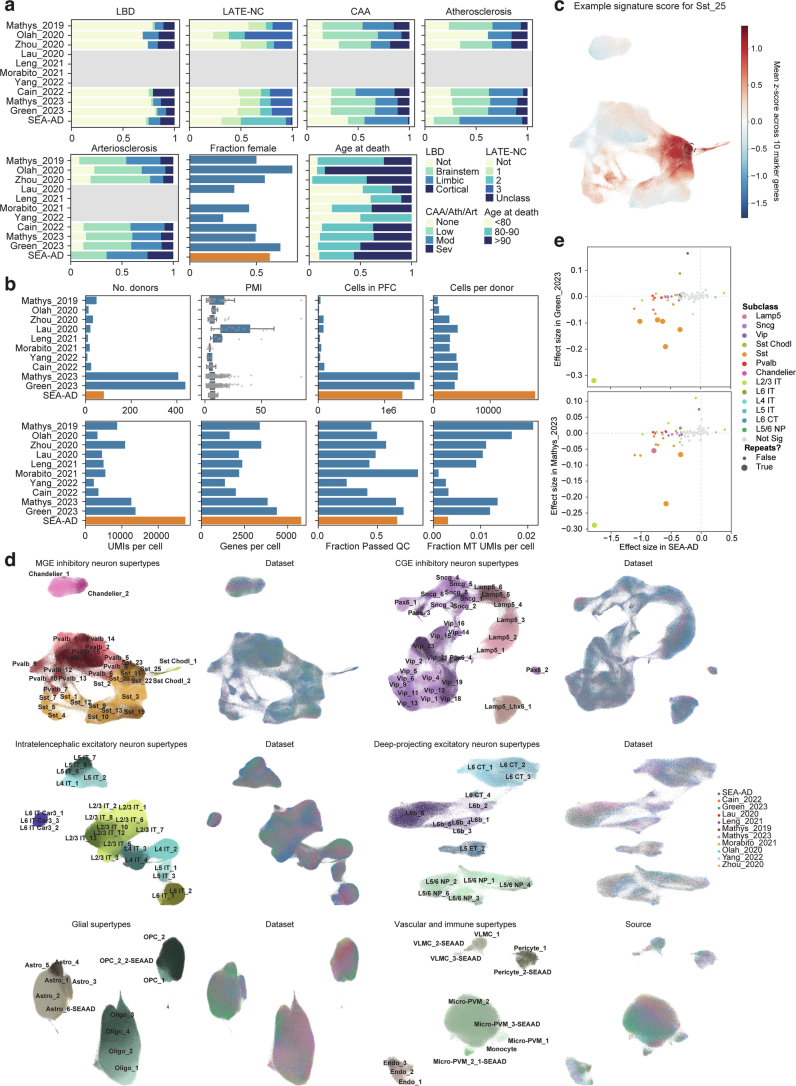
Update of
-
Integrated multimodal cell atlas of Alzheimer's disease.Res Sq [Preprint]. 2023 May 23:rs.3.rs-2921860. doi: 10.21203/rs.3.rs-2921860/v1. Res Sq. 2023. Update in: Nat Neurosci. 2024 Dec;27(12):2366-2383. doi: 10.1038/s41593-024-01774-5. PMID: 37292694 Free PMC article. Updated. Preprint.
References
MeSH terms
Grants and funding
- P30 AG072975/AG/NIA NIH HHS/United States
- U24 NS135561/NS/NINDS NIH HHS/United States
- R01 AG066831/AG/NIA NIH HHS/United States
- RF1 AG062377/AG/NIA NIH HHS/United States
- T32 GM087237/GM/NIGMS NIH HHS/United States
- U01AG006781/U.S. Department of Health & Human Services | NIH | National Institute on Aging (U.S. National Institute on Aging)
- U01 AG006781/AG/NIA NIH HHS/United States
- RF1 AG057473/AG/NIA NIH HHS/United States
- P50AG005136/U.S. Department of Health & Human Services | NIH | National Institute on Aging (U.S. National Institute on Aging)
- U19 AG066567/AG/NIA NIH HHS/United States
- UM1 MH130981/MH/NIMH NIH HHS/United States
- RF1 AG054012/AG/NIA NIH HHS/United States
- U19AG066567/U.S. Department of Health & Human Services | NIH | National Institute on Aging (U.S. National Institute on Aging)
- U01 AG046152/AG/NIA NIH HHS/United States
- R01 AG058002/AG/NIA NIH HHS/United States
- R01 HG008155/HG/NHGRI NIH HHS/United States
- R01 NS105984/NS/NINDS NIH HHS/United States
- U24 NS133949/NS/NINDS NIH HHS/United States
- U01 AG061356/AG/NIA NIH HHS/United States
- UG3 NS115064/NS/NINDS NIH HHS/United States
- U01 AG032984/AG/NIA NIH HHS/United States
- R01 AG030146/AG/NIA NIH HHS/United States
- R01 AG060942/AG/NIA NIH HHS/United States
- U24 NS133945/NS/NINDS NIH HHS/United States
- U01 AG072572/AG/NIA NIH HHS/United States
- R01 AG017917/AG/NIA NIH HHS/United States
- UH3 NS115064/NS/NINDS NIH HHS/United States
- R01 AG074003/AG/NIA NIH HHS/United States
- P50 AG005136/AG/NIA NIH HHS/United States
- RF1 AG065406/AG/NIA NIH HHS/United States
- R01 AG036042/AG/NIA NIH HHS/United States
- P30 AG010161/AG/NIA NIH HHS/United States
- R01 NS127187/NS/NINDS NIH HHS/United States
- U19 AG060909/AG/NIA NIH HHS/United States
- P30 AG066509/AG/NIA NIH HHS/United States
- RC2 AG036547/AG/NIA NIH HHS/United States
- RF1 AG054321/AG/NIA NIH HHS/United States
- U19AG060909/U.S. Department of Health & Human Services | NIH | National Institute on Aging (U.S. National Institute on Aging)
- U01 AG046161/AG/NIA NIH HHS/United States
- U01 NS110453/NS/NINDS NIH HHS/United States
- R01 AG048015/AG/NIA NIH HHS/United States
- R01 AG062335/AG/NIA NIH HHS/United States
- P30AG066509/U.S. Department of Health & Human Services | NIH | National Institute on Aging (U.S. National Institute on Aging)
- U24 AG072458/AG/NIA NIH HHS/United States
- U01 MH119509/MH/NIMH NIH HHS/United States
- R01 AG036836/AG/NIA NIH HHS/United States
- R01 AG015819/AG/NIA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
