

SDePER: a hybrid machine learning and regression method for cell-type deconvolution of spatial barcoding-based transcriptomic data
- PMID: 39402626
- PMCID: PMC11475911
- DOI: 10.1186/s13059-024-03416-2
SDePER: a hybrid machine learning and regression method for cell-type deconvolution of spatial barcoding-based transcriptomic data
Abstract
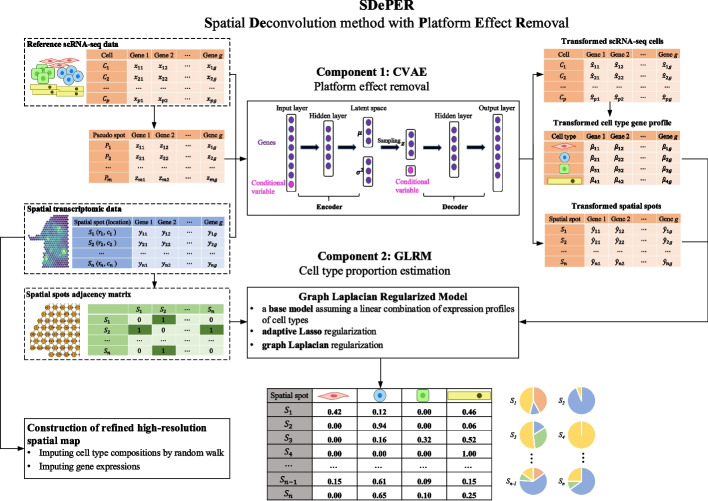
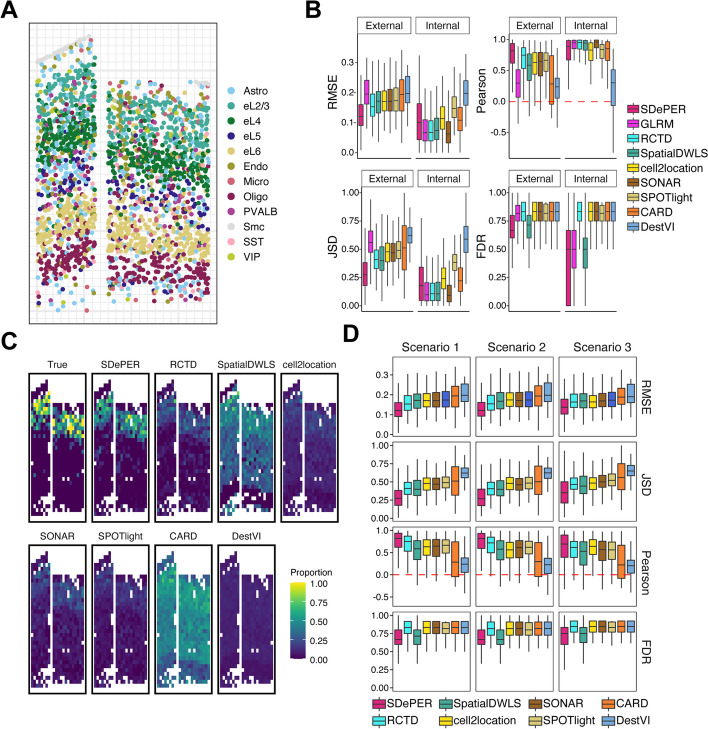
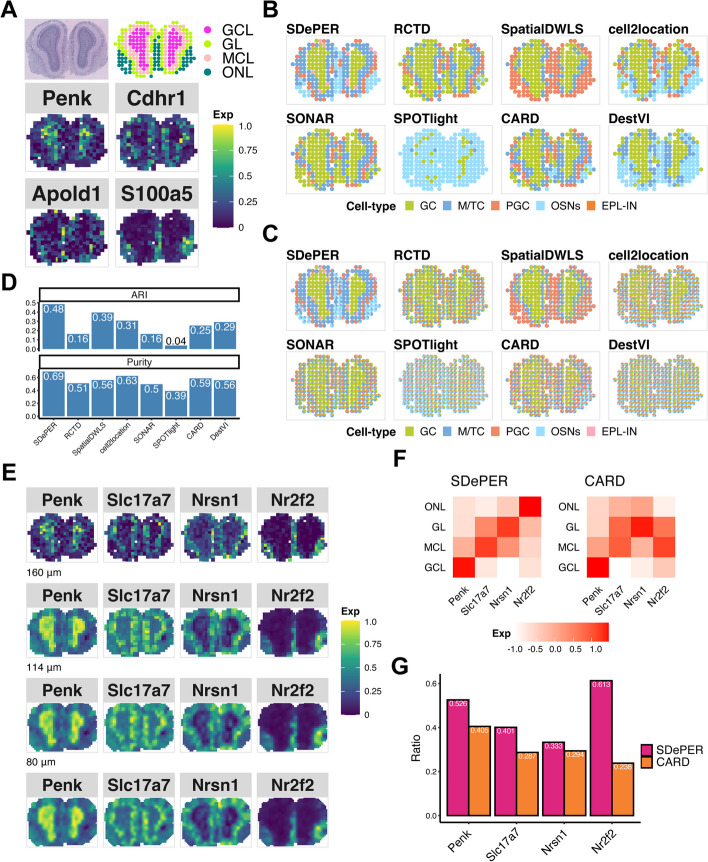
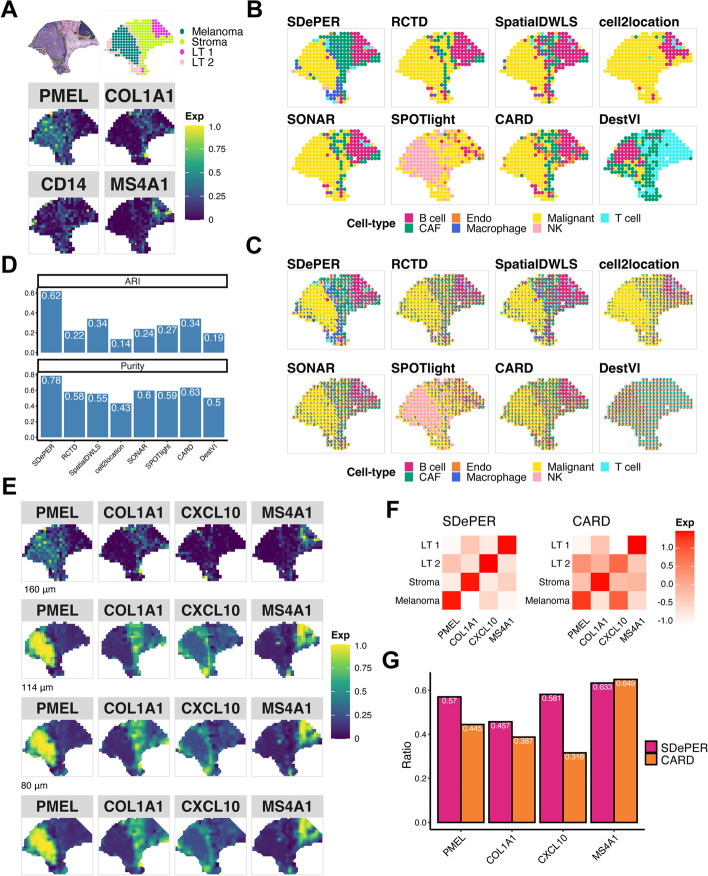
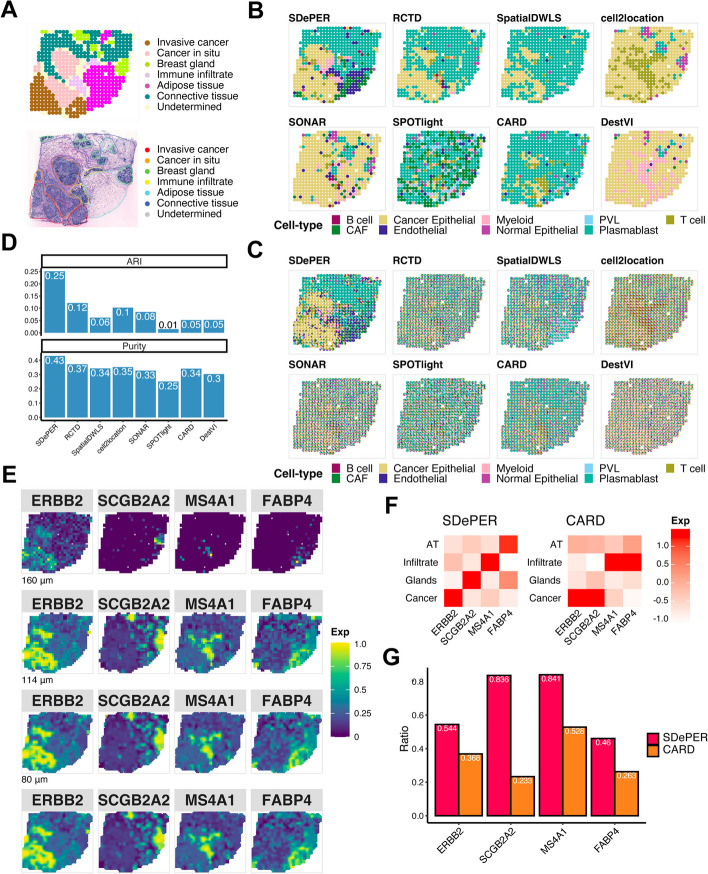
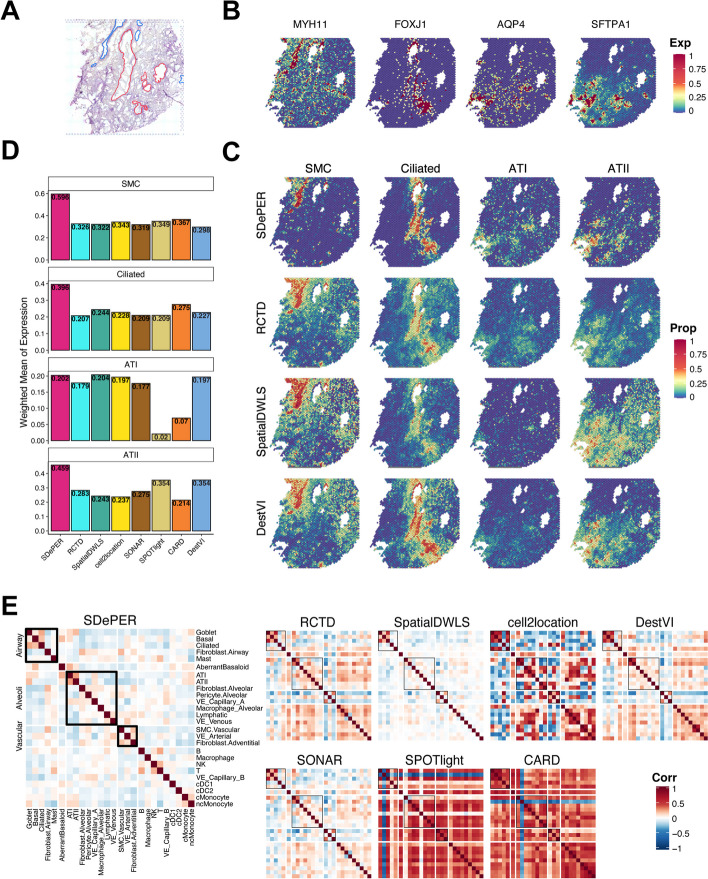
Spatial barcoding-based transcriptomic (ST) data require deconvolution for cellular-level downstream analysis. Here we present SDePER, a hybrid machine learning and regression method to deconvolve ST data using reference single-cell RNA sequencing (scRNA-seq) data. SDePER tackles platform effects between ST and scRNA-seq data, ensuring a linear relationship between them while addressing sparsity and spatial correlations in cell types across capture spots. SDePER estimates cell-type proportions, enabling enhanced resolution tissue mapping by imputing cell-type compositions and gene expressions at unmeasured locations. Applications to simulated data and four real datasets showed SDePER's superior accuracy and robustness over existing methods.
© 2024. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Update of
-
A hybrid machine learning and regression method for cell type deconvolution of spatial barcoding-based transcriptomic data.bioRxiv [Preprint]. 2023 Aug 26:2023.08.24.554722. doi: 10.1101/2023.08.24.554722. bioRxiv. 2023. Update in: Genome Biol. 2024 Oct 14;25(1):271. doi: 10.1186/s13059-024-03416-2. PMID: 37662370 Free PMC article. Updated. Preprint.
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
