

When less is more: sketching with minimizers in genomics
- PMID: 39402664
- PMCID: PMC11472564
- DOI: 10.1186/s13059-024-03414-4
When less is more: sketching with minimizers in genomics
Abstract
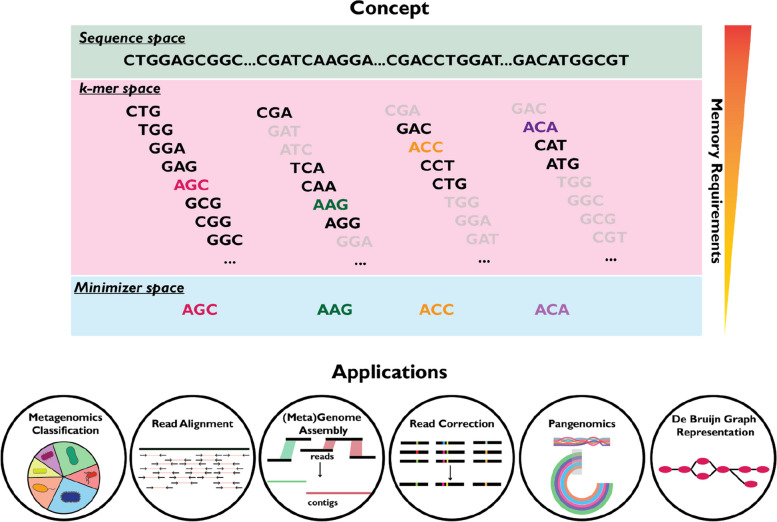
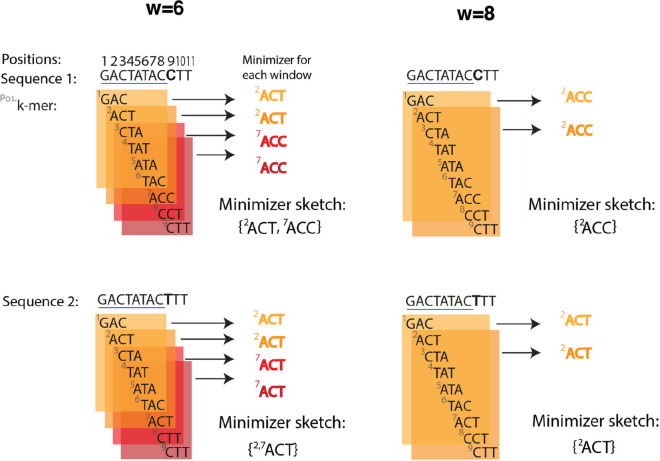
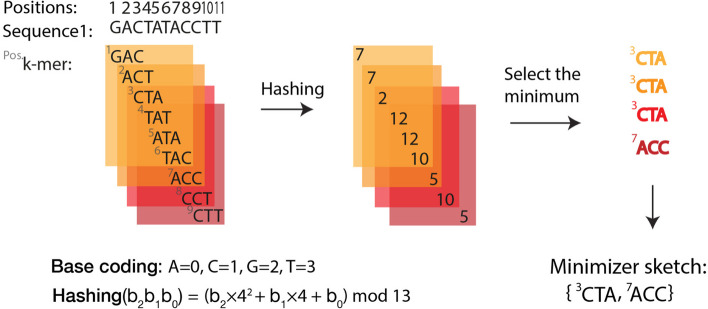
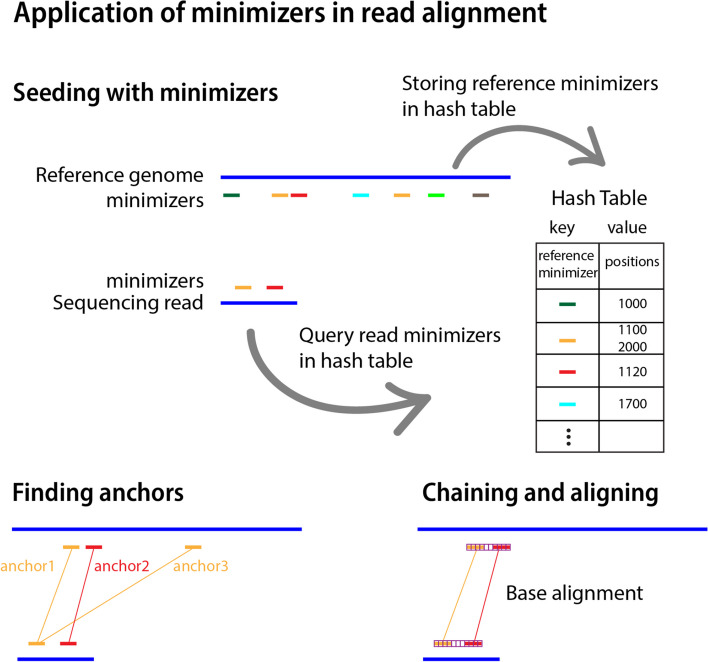
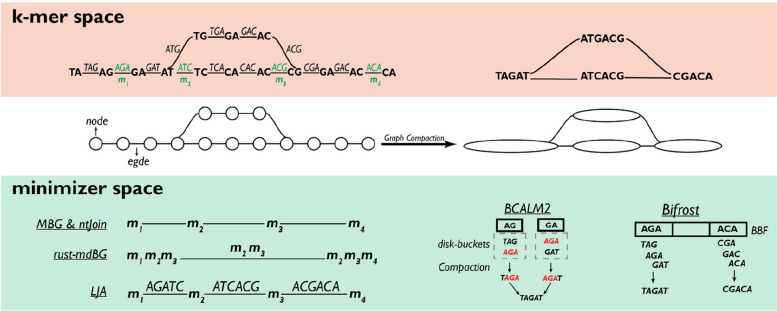
The exponential increase in sequencing data calls for conceptual and computational advances to extract useful biological insights. One such advance, minimizers, allows for reducing the quantity of data handled while maintaining some of its key properties. We provide a basic introduction to minimizers, cover recent methodological developments, and review the diverse applications of minimizers to analyze genomic data, including de novo genome assembly, metagenomics, read alignment, read correction, and pangenomes. We also touch on alternative data sketching techniques including universal hitting sets, syncmers, or strobemers. Minimizers and their alternatives have rapidly become indispensable tools for handling vast amounts of data.
© 2024. The Author(s).
Conflict of interest statement
FJS received support from PacBio, ONT, and Illumina. CD has been providing consulting services for Pacific Biosciences, Inc. All other authors declare that they have no competing interests.
Figures
References
-
- Sunagawa S, Acinas SG, Bork P, Bowler C, Tara Oceans Coordinators, Eveillard D, et al. Tara Oceans: towards global ocean ecosystems biology. Nat Rev Microbiol. 2020;18:428–45. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
