

Employing Multi-Omics Analyses to Understand Changes during Kidney Development in Perinatal Interleukin-6 Animal Model
- PMID: 39404429
- PMCID: PMC11476440
- DOI: 10.3390/cells13191667
Employing Multi-Omics Analyses to Understand Changes during Kidney Development in Perinatal Interleukin-6 Animal Model
Abstract
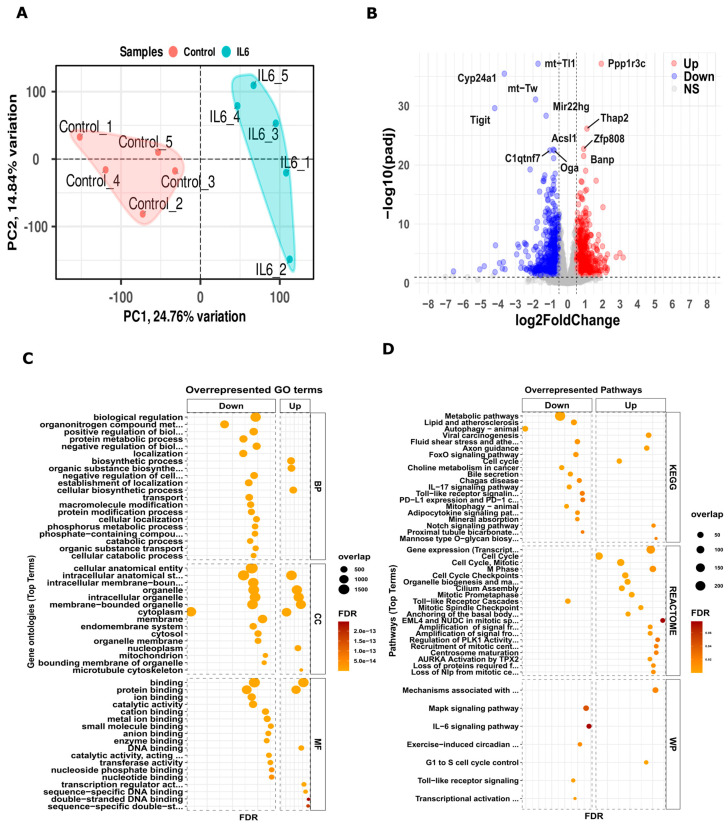
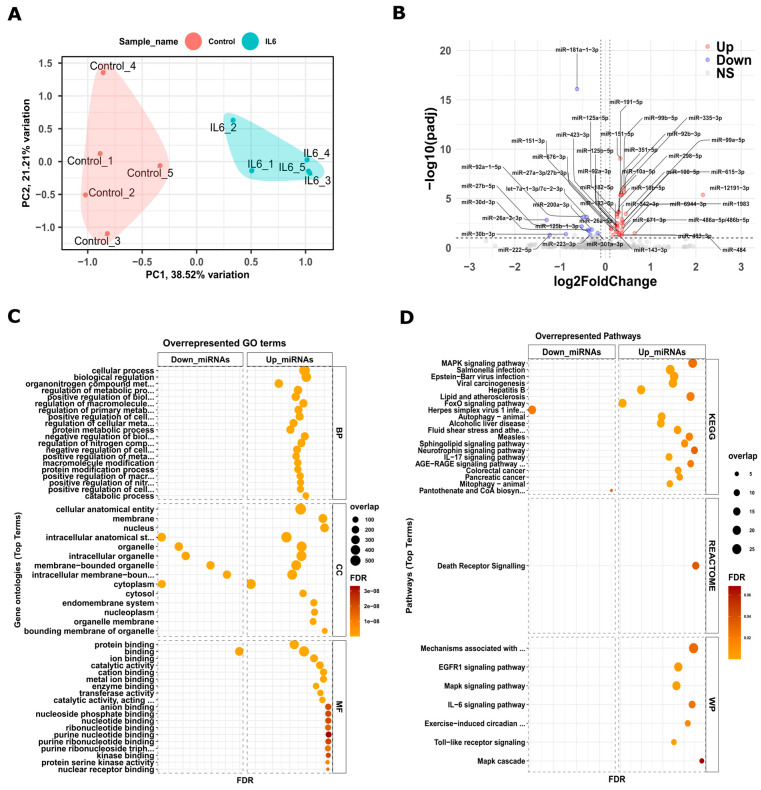
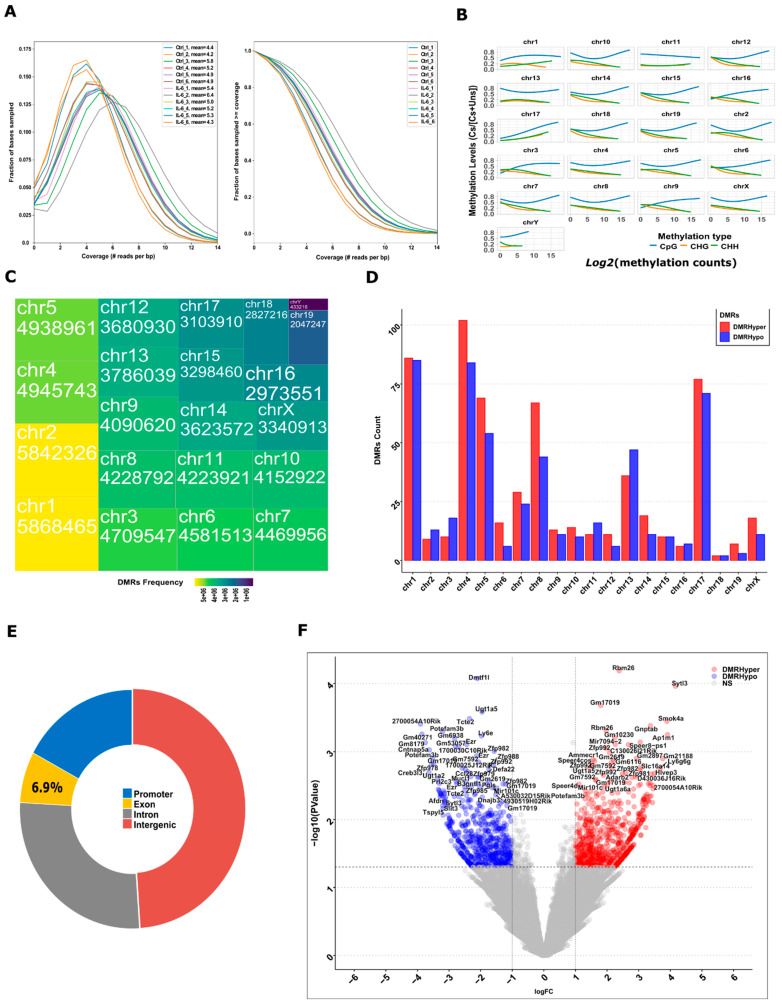
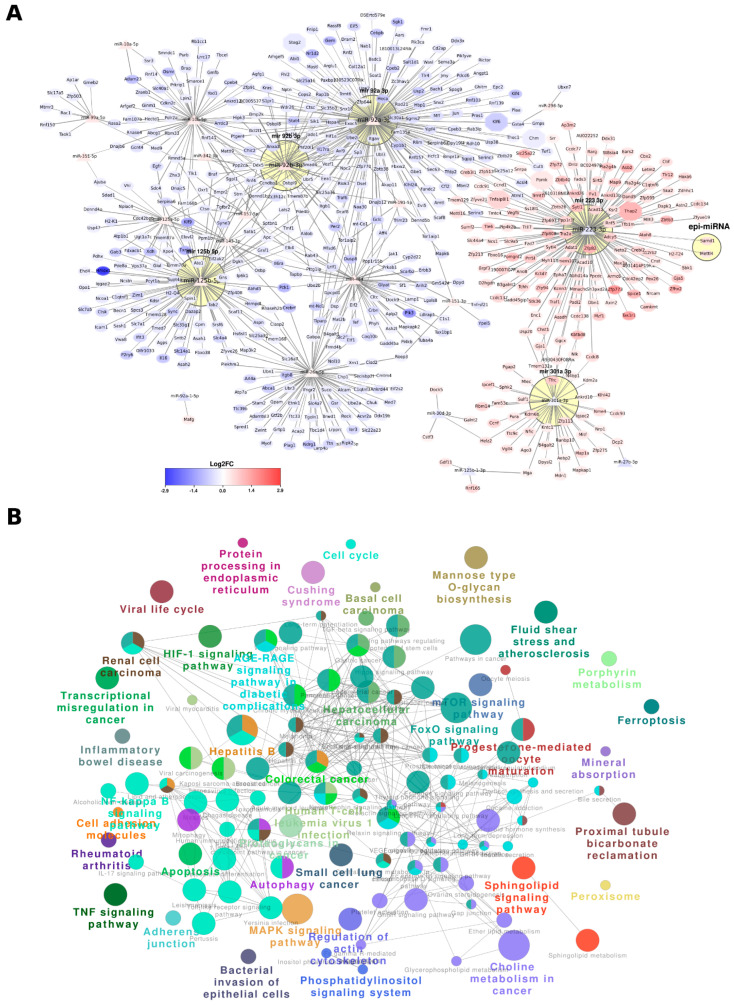
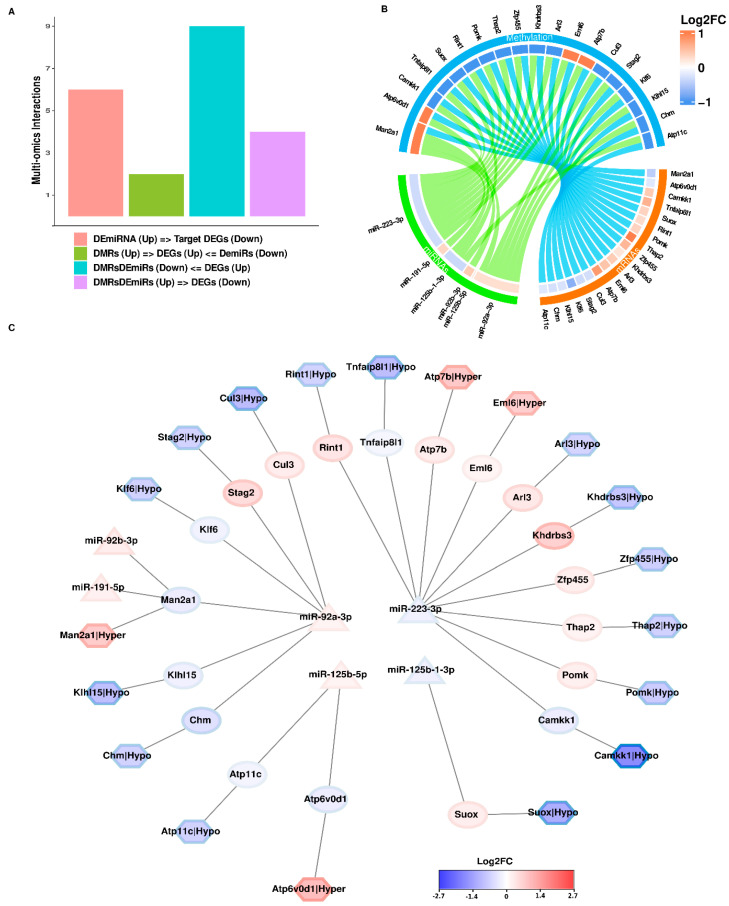
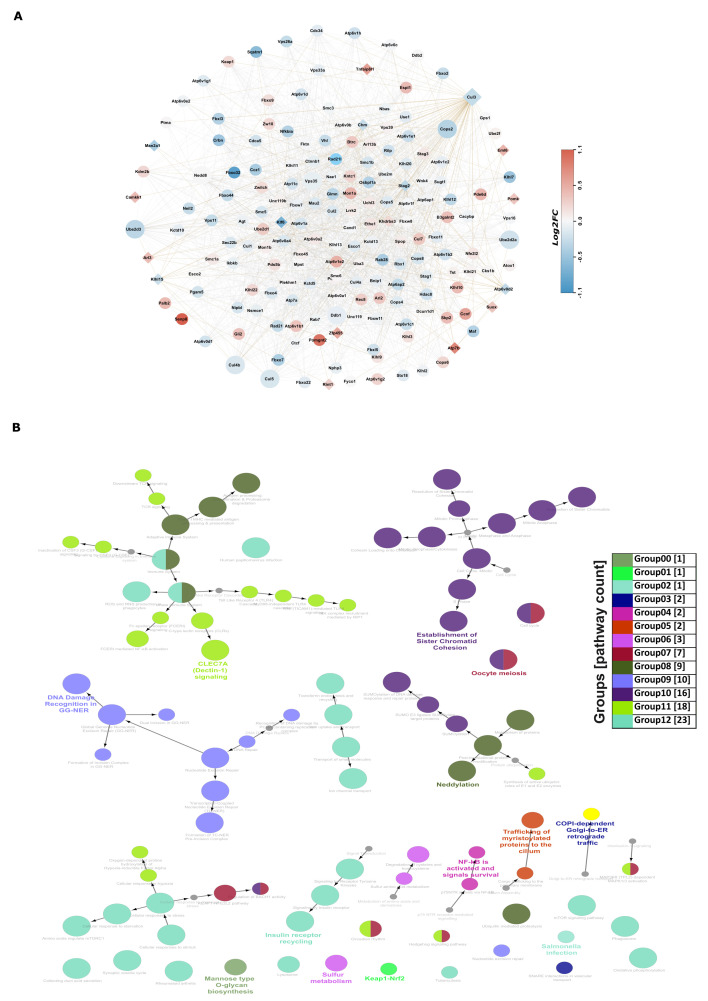
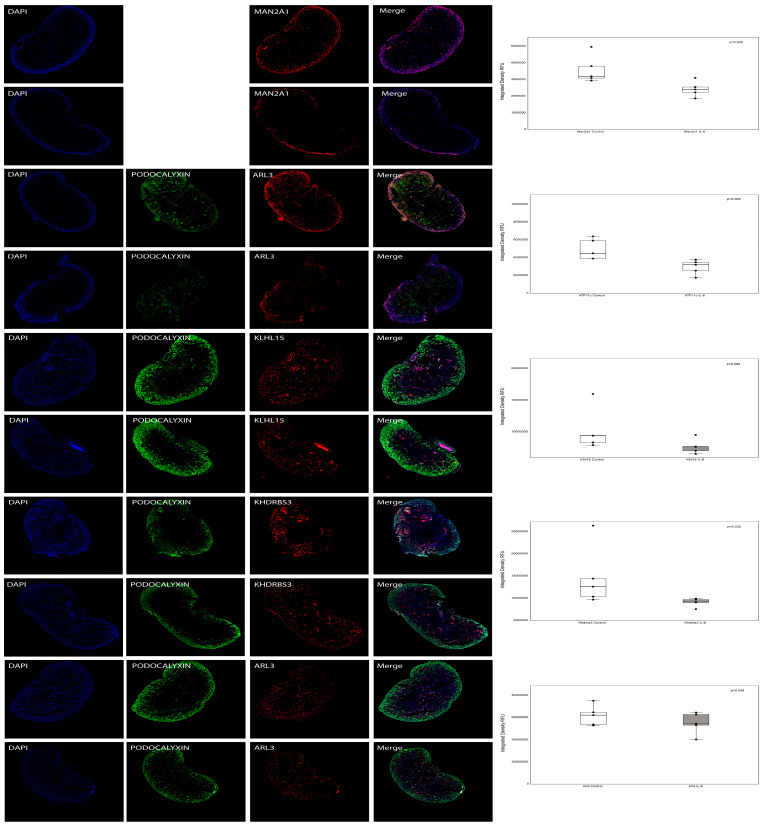
Chronic kidney disease (CKD) is a leading cause of morbidity and mortality globally. Maternal obesity during pregnancy is linked to systemic inflammation and elevated levels of the pro-inflammatory cytokine interleukin-6 (IL-6). In our previous work, we demonstrated that increased maternal IL-6 during gestation impacts intrauterine development in mice. We hypothesized that IL-6-induced inflammation alters gene expression in the developing fetus. To test this, pregnant mice were administered IL-6 or saline during mid-gestation. Newborn mouse kidneys were analyzed using mRNA-seq, miRNA-seq and whole-genome bisulfite-seq (WGBS). A multi-omics approach was employed to quantify mRNA gene expression, miRNA expression and DNA methylation, using advanced bioinformatics and data integration techniques. Our analysis identified 19 key genes present in multiple omics datasets, regulated by epigenetics and miRNAs. We constructed a regulatory network for these genes, revealing disruptions in pathways such as Mannose type O-glycan biosynthesis, the cell cycle, apoptosis and FoxO signaling. Notably, the Atp7b gene was regulated by DNA methylation and miR-223 targeting, whereas the Man2a1 gene was controlled by DNA methylation affecting energy metabolism. These findings suggest that these genes may play a role in fetal programming, potentially leading to CKD later in life due to gestational inflammation.
Keywords: chronic kidney disease (CKD); co-expression; epigenetics; interleukin-6 (IL-6); miRNA regulation; multi-omics.
Conflict of interest statement
The authors have declared no conflicts of interest.
Figures
References
-
- Lee Y.Q., Lumbers E.R., Oldmeadow C., Collins C.E., Johnson V., Keogh L., Sutherland K., Gordon A., Smith R., Rae K.M., et al. The Relationship between Maternal Adiposity during Pregnancy and Fetal Kidney Development and Kidney Function in Infants: The Gomeroi Gaaynggal Study. Physiol. Rep. 2019;7:e14227. doi: 10.14814/phy2.14227. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
