

Long-read sequencing of an advanced cancer cohort resolves rearrangements, unravels haplotypes, and reveals methylation landscapes
- PMID: 39406235
- PMCID: PMC11605692
- DOI: 10.1016/j.xgen.2024.100674
Long-read sequencing of an advanced cancer cohort resolves rearrangements, unravels haplotypes, and reveals methylation landscapes
Abstract
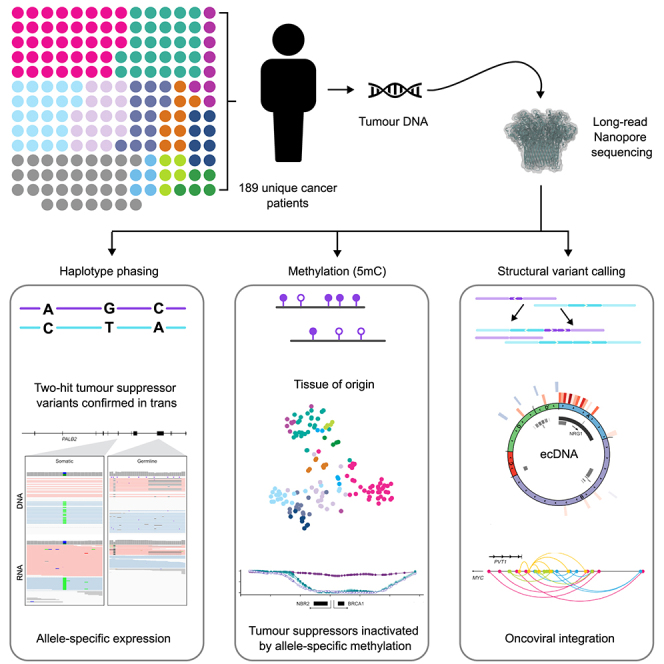
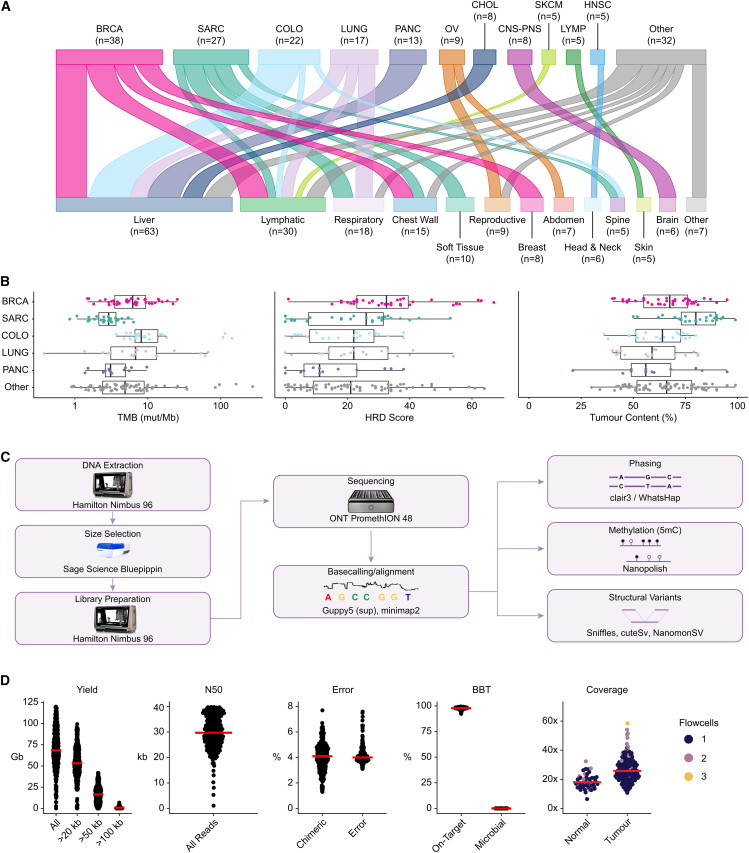
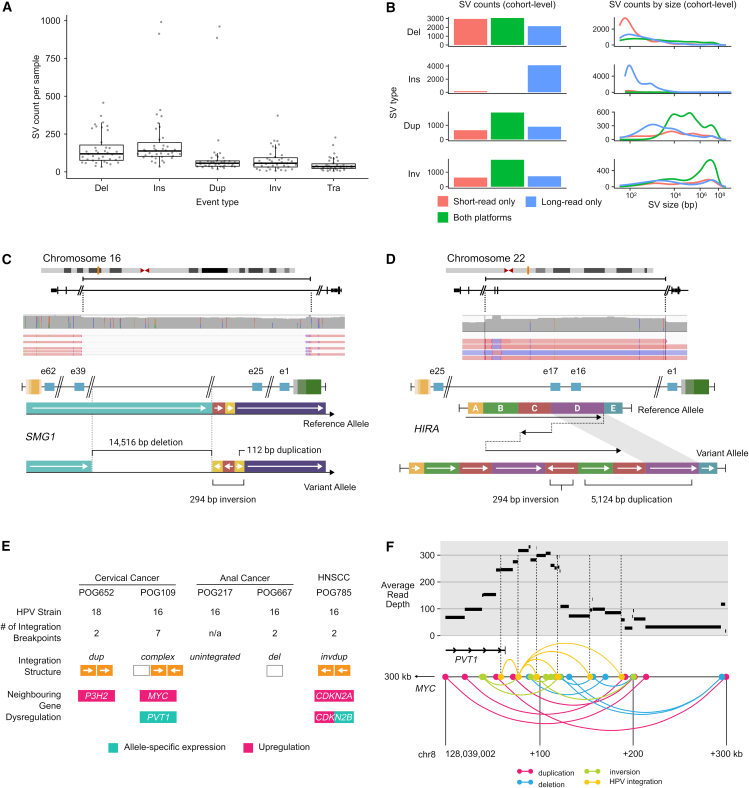
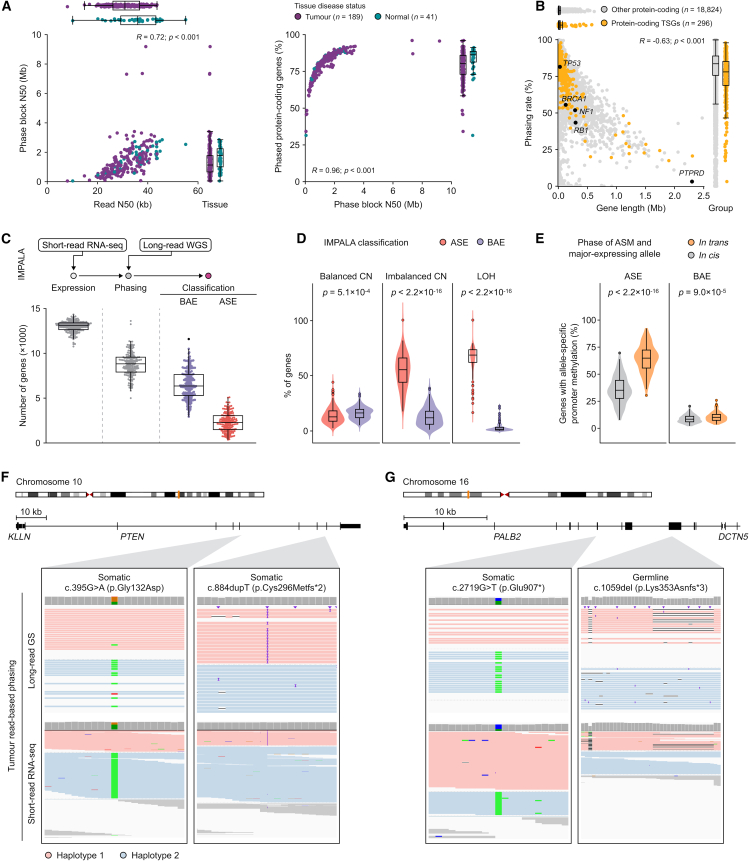
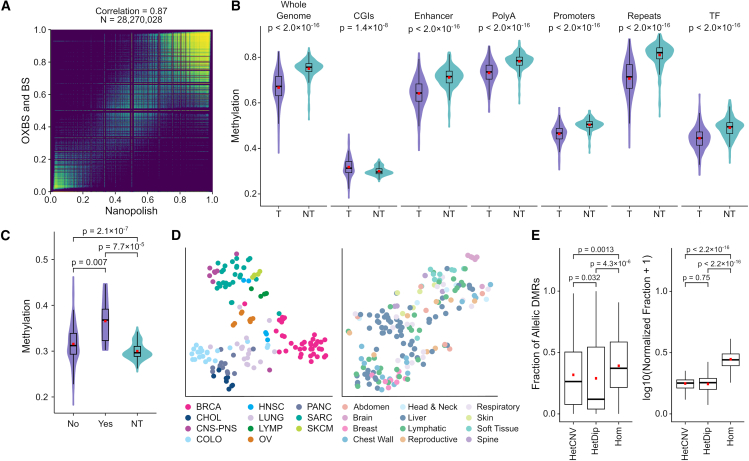
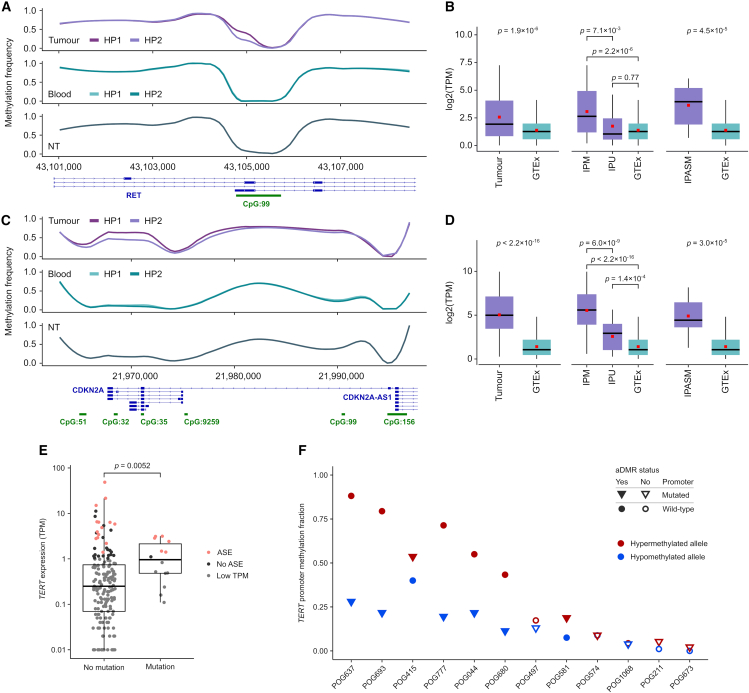
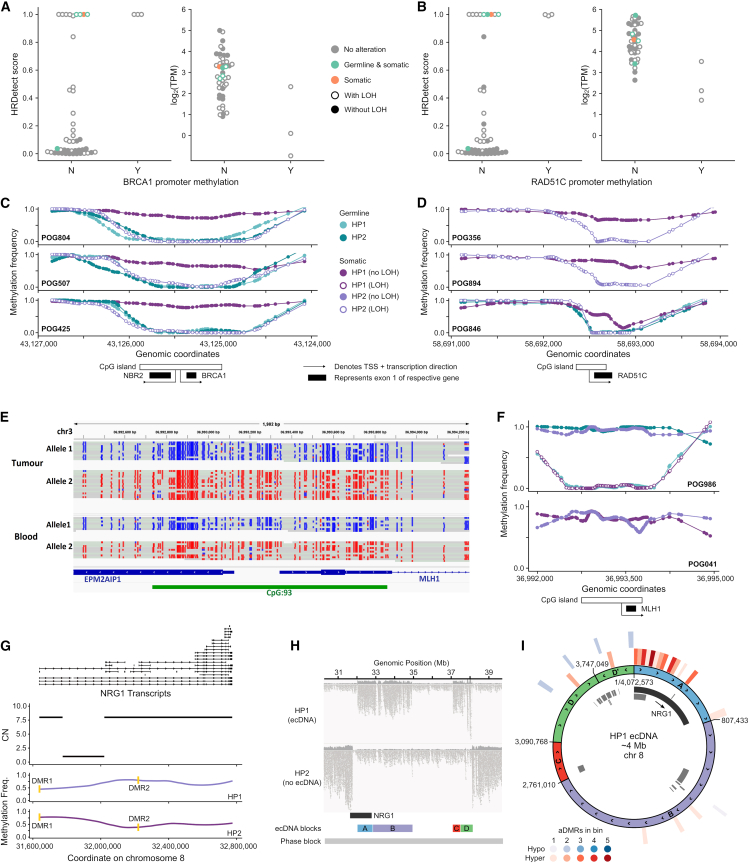
The Long-Read Personalized OncoGenomics (POG) dataset comprises a cohort of 189 patient tumors and 41 matched normal samples sequenced using the Oxford Nanopore Technologies PromethION platform. This dataset from the POG program and the Marathon of Hope Cancer Centres Network includes DNA and RNA short-read sequence data, analytics, and clinical information. We show the potential of long-read sequencing for resolving complex cancer-related structural variants, viral integrations, and extrachromosomal circular DNA. Long-range phasing facilitates the discovery of allelically differentially methylated regions (aDMRs) and allele-specific expression, including recurrent aDMRs in the cancer genes RET and CDKN2A. Germline promoter methylation in MLH1 can be directly observed in Lynch syndrome. Promoter methylation in BRCA1 and RAD51C is a likely driver behind homologous recombination deficiency where no coding driver mutation was found. This dataset demonstrates applications for long-read sequencing in precision medicine and is available as a resource for developing analytical approaches using this technology.
Keywords: TFRI MOHCCN; allele-specific expression; allelically differentially methylated regions (aDMRs); cancer genomics; extrachromosomal DNA; homologous recombination deficiency; long-range phasing; nanopore long-read sequencing; personalized medicine; structural variant detection.
Copyright © 2024 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The following authors disclose relevant potential competing interests: K.O.N., V.P., L.F.P., K.D., J.L., and S.J.M.J. received travel funding from Oxford Nanopore Technologies to present at conferences in 2022 and 2023.
Figures
References
-
- Pleasance E., Titmuss E., Williamson L., Kwan H., Culibrk L., Zhao E.Y., Dixon K., Fan K., Bowlby R., Jones M.R., et al. Pan-cancer analysis of advanced patient tumors reveals interactions between therapy and genomic landscapes. Nat. Cancer. 2020;1:452–468. doi: 10.1038/s43018-020-0050-6. - DOI - PubMed
-
- Wong M., Mayoh C., Lau L.M.S., Khuong-Quang D.-A., Pinese M., Kumar A., Barahona P., Wilkie E.E., Sullivan P., Bowen-James R., et al. Whole genome, transcriptome and methylome profiling enhances actionable target discovery in high-risk pediatric cancer. Nat. Med. 2020;26:1742–1753. doi: 10.1038/s41591-020-1072-4. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
