

Seamless, rapid, and accurate analyses of outbreak genomic data using split k-mer analysis
- PMID: 39406504
- PMCID: PMC11529842
- DOI: 10.1101/gr.279449.124
Seamless, rapid, and accurate analyses of outbreak genomic data using split k-mer analysis
Abstract
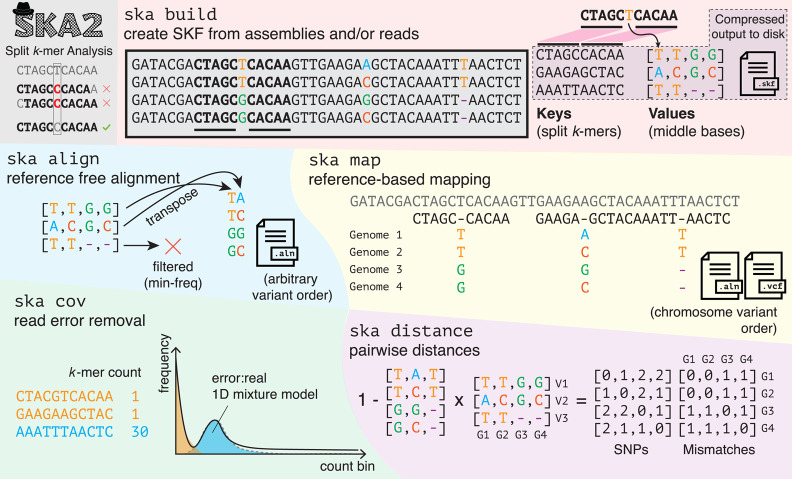
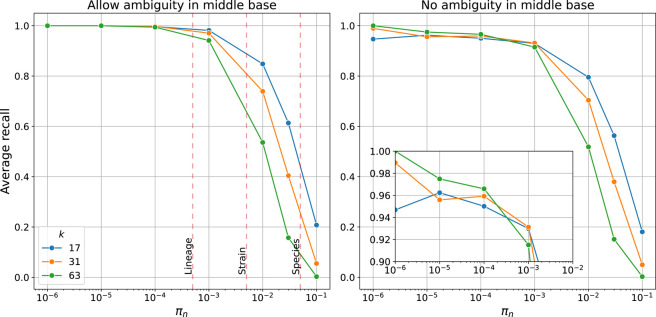
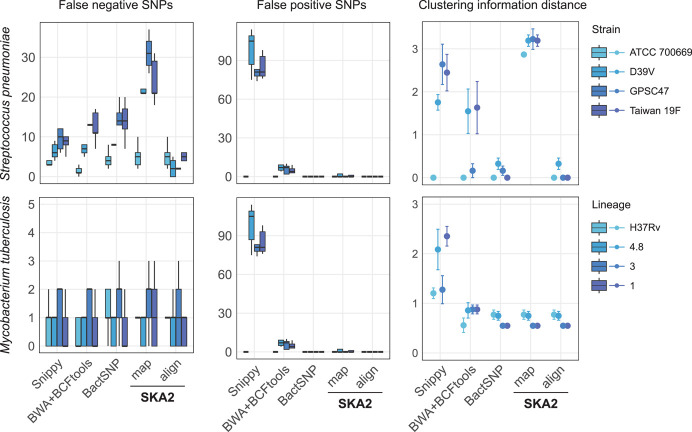
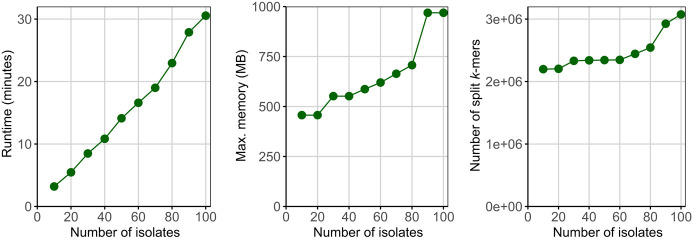
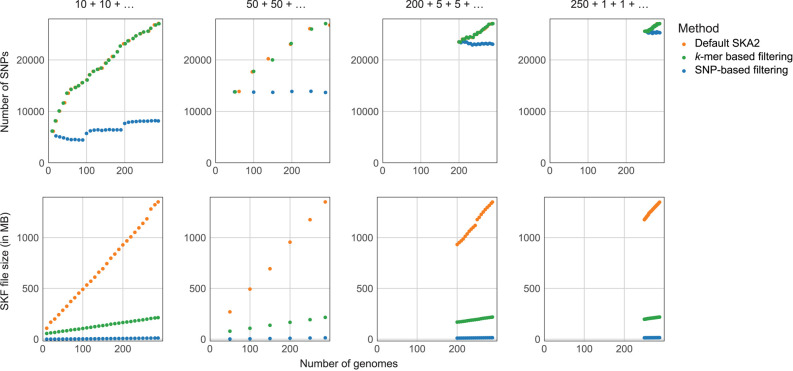
Sequence variation observed in populations of pathogens can be used for important public health and evolutionary genomic analyses, especially outbreak analysis and transmission reconstruction. Identifying this variation is typically achieved by aligning sequence reads to a reference genome, but this approach is susceptible to reference biases and requires careful filtering of called genotypes. There is a need for tools that can process this growing volume of bacterial genome data, providing rapid results, but that remain simple so they can be used without highly trained bioinformaticians, expensive data analysis, and long-term storage and processing of large files. Here we describe split k-mer analysis (SKA2), a method that supports both reference-free and reference-based mapping to quickly and accurately genotype populations of bacteria using sequencing reads or genome assemblies. SKA2 is highly accurate for closely related samples, and in outbreak simulations, we show superior variant recall compared with reference-based methods, with no false positives. SKA2 can also accurately map variants to a reference and be used with recombination detection methods to rapidly reconstruct vertical evolutionary history. SKA2 is many times faster than comparable methods and can be used to add new genomes to an existing call set, allowing sequential use without the need to reanalyze entire collections. With an inherent absence of reference bias, high accuracy, and a robust implementation, SKA2 has the potential to become the tool of choice for genotyping bacteria. SKA2 is implemented in Rust and is freely available as open-source software.
© 2024 Derelle et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Becker HEF, Jamin C, Bervoets L, Boleij A, Xu P, Pierik MJ, Stassen FRM, Savelkoul PHM, Penders J, Jonkers DMAE. 2021. Higher prevalence of Bacteroides fragilis in Crohn's disease exacerbations and strain-dependent increase of epithelial resistance. Front Microbiol 12: 598232. 10.3389/fmicb.2021.598232 - DOI - PMC - PubMed
-
- Bickhart DM, Kolmogorov M, Tseng E, Portik DM, Korobeynikov A, Tolstoganov I, Uritskiy G, Liachko I, Sullivan ST, Shin SB, et al. 2022. Generating lineage-resolved, complete metagenome-assembled genomes from complex microbial communities. Nat Biotechnol 40: 711–719. 10.1038/s41587-021-01130-z - DOI - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous