

USP7 depletion potentiates HIF2α degradation and inhibits clear cell renal cell carcinoma progression
- PMID: 39406703
- PMCID: PMC11482519
- DOI: 10.1038/s41419-024-07136-0
USP7 depletion potentiates HIF2α degradation and inhibits clear cell renal cell carcinoma progression
Abstract
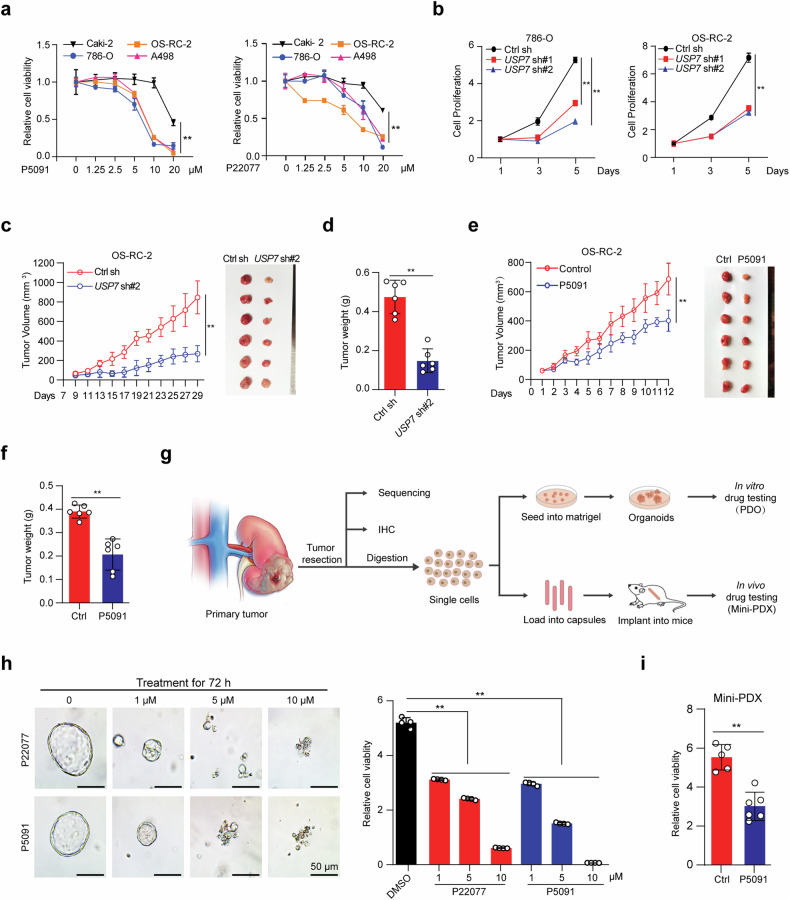
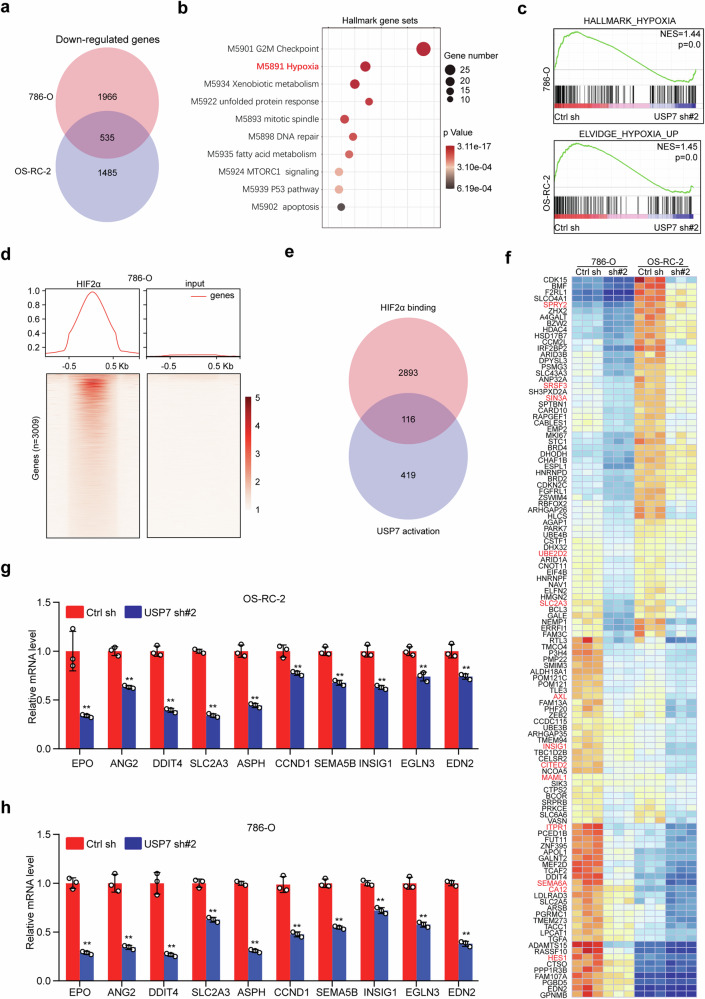
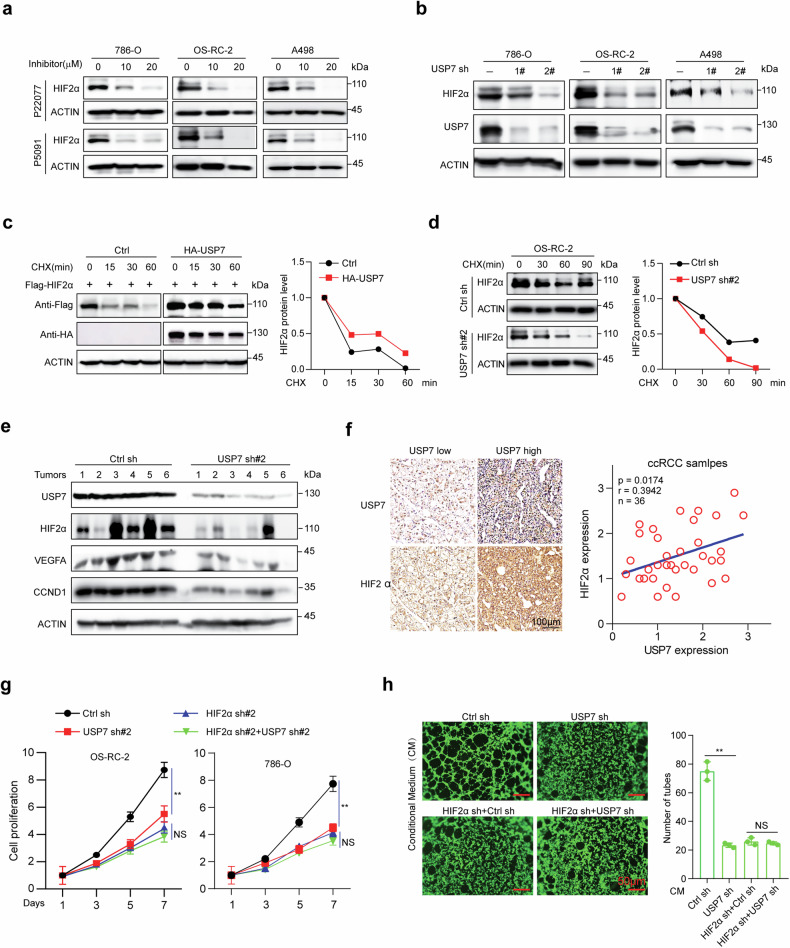
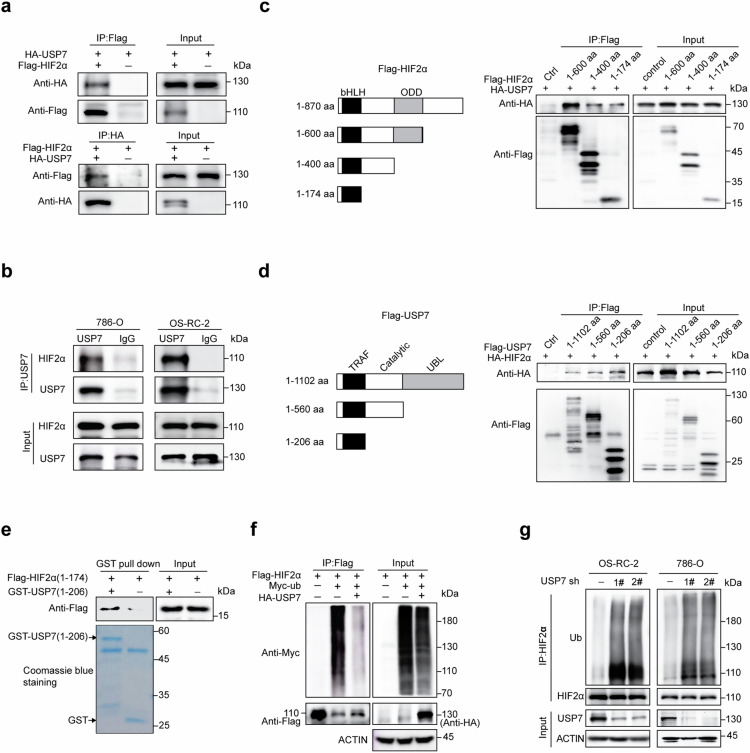
Clear cell renal cell carcinoma (ccRCC) is characterized by Von Hippel Lindau (VHL) gene loss of function mutation, which leads to the accumulation of hypoxia-inducible factor 2α (HIF2α). HIF2α has been well-established as one of the major oncogenic drivers of ccRCC, however, its therapeutic targeting remains a challenge. Through an analysis of proteomic data from ccRCCs and adjacent non-tumor tissues, we herein revealed that Ubiquitin-Specific Peptidase 7 (USP7) was upregulated in tumor tissues, and its depletion by inhibitors or shRNAs caused significant suppression of tumor progression in vitro and in vivo. Mechanistically, USP7 expression is activated by the transcription factors FUBP1 and FUBP3, and it promotes tumor progression mainly by deubiquitinating and stabilizing HIF2α. Moreover, the combination of USP7 inhibitors and afatinib (an ERBB family inhibitor) coordinately induce cell death and tumor suppression. In mechanism, afatinib indirectly inhibits USP7 transcription and accelerates the degradation of HIF2α protein, and the combination of them caused a more profound suppression of HIF2α abundance. These findings reveal a FUBPs-USP7-HIF2α regulatory axis that underlies the progression of ccRCC and provides a rationale for therapeutic targeting of oncogenic HIF2α via combinational treatment of USP7 inhibitor and afatinib.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

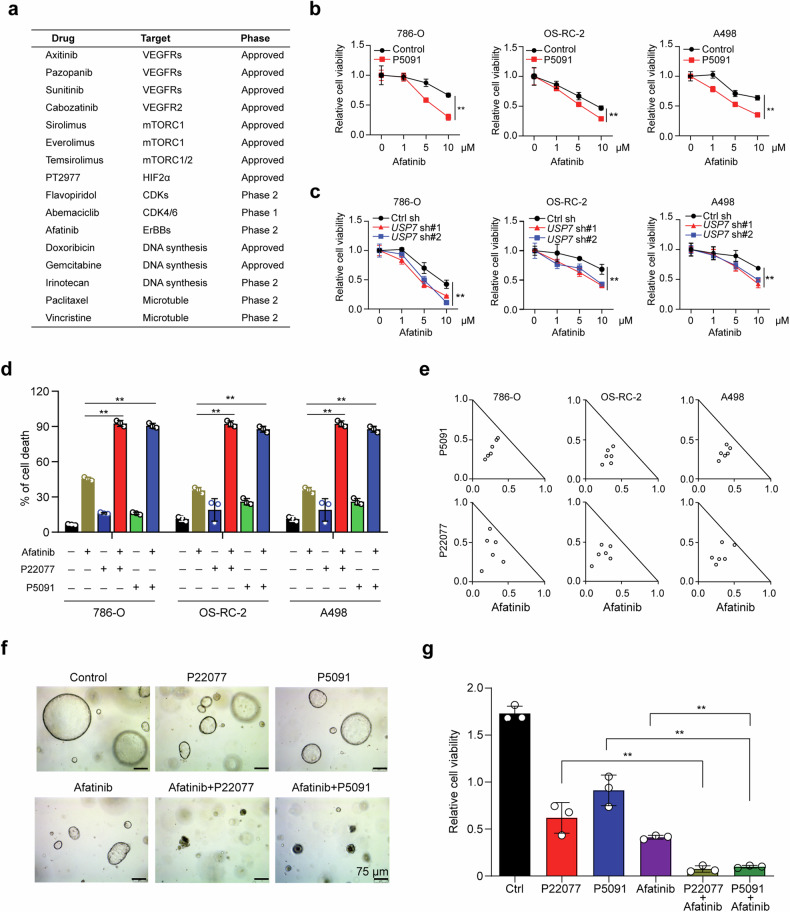
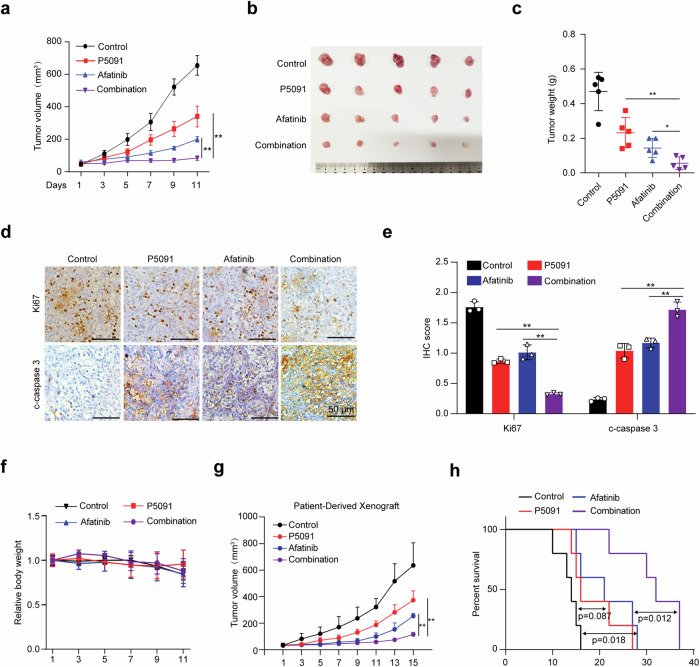
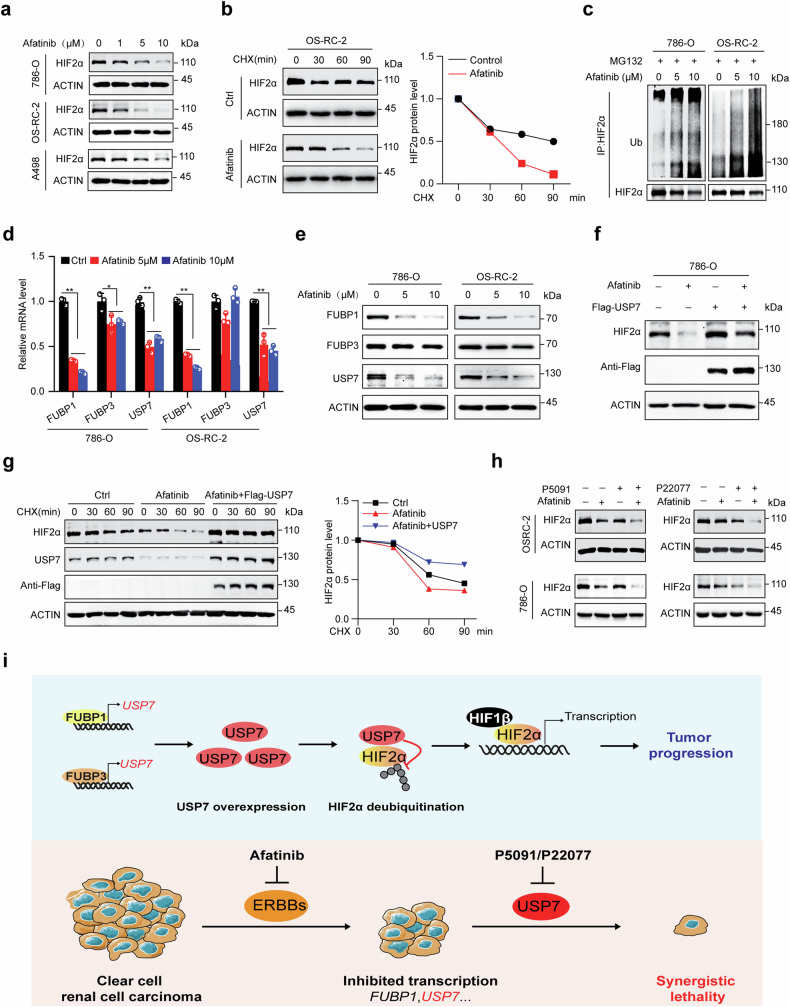
References
-
- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. - PubMed
-
- Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, et al. Targeting of HIF-alpha to the von Hippel–Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–72. - PubMed
-
- Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–8. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
