

A cross-disease resource of living human microglia identifies disease-enriched subsets and tool compounds recapitulating microglial states
- PMID: 39406950
- PMCID: PMC12094270
- DOI: 10.1038/s41593-024-01764-7
A cross-disease resource of living human microglia identifies disease-enriched subsets and tool compounds recapitulating microglial states
Abstract
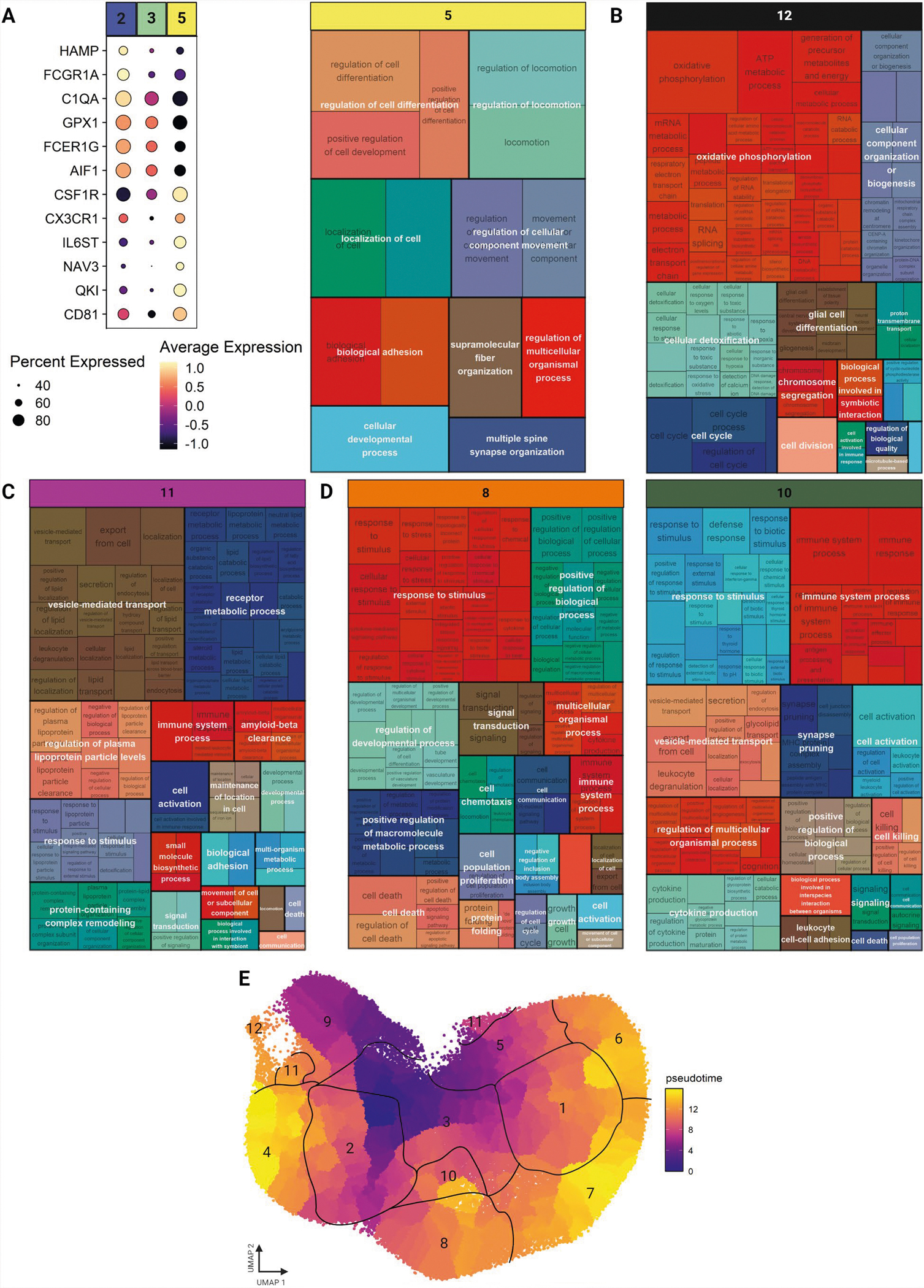
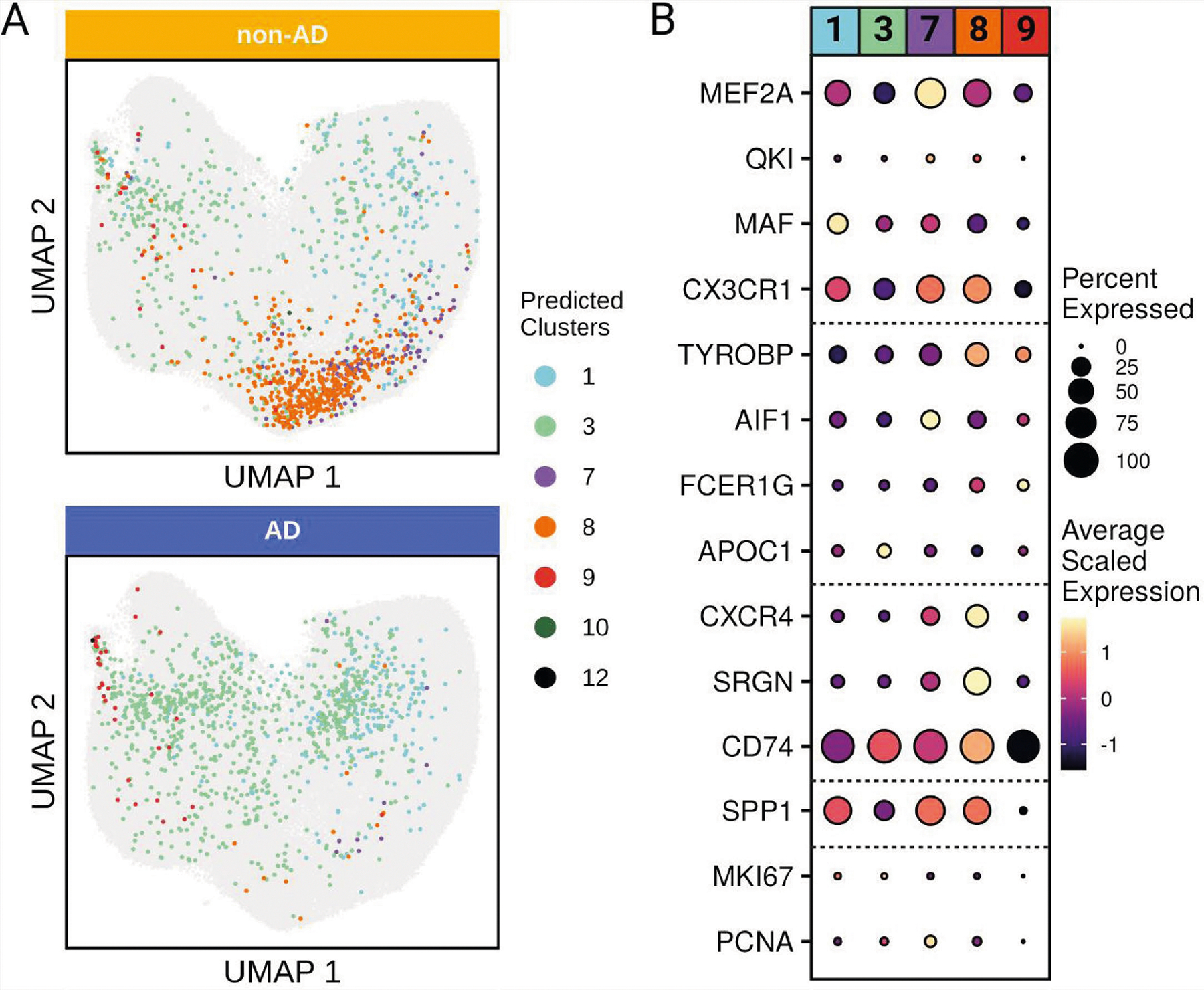
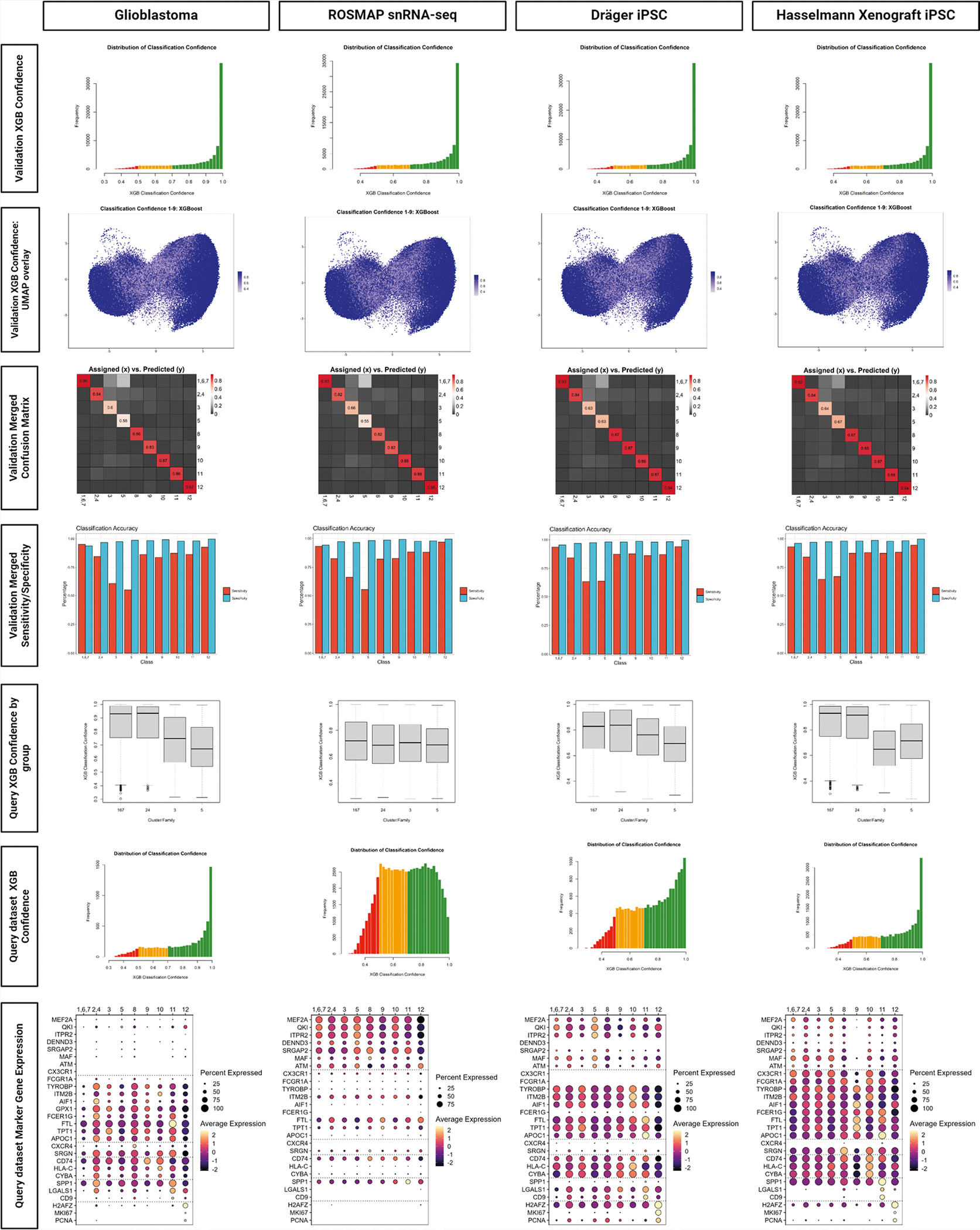
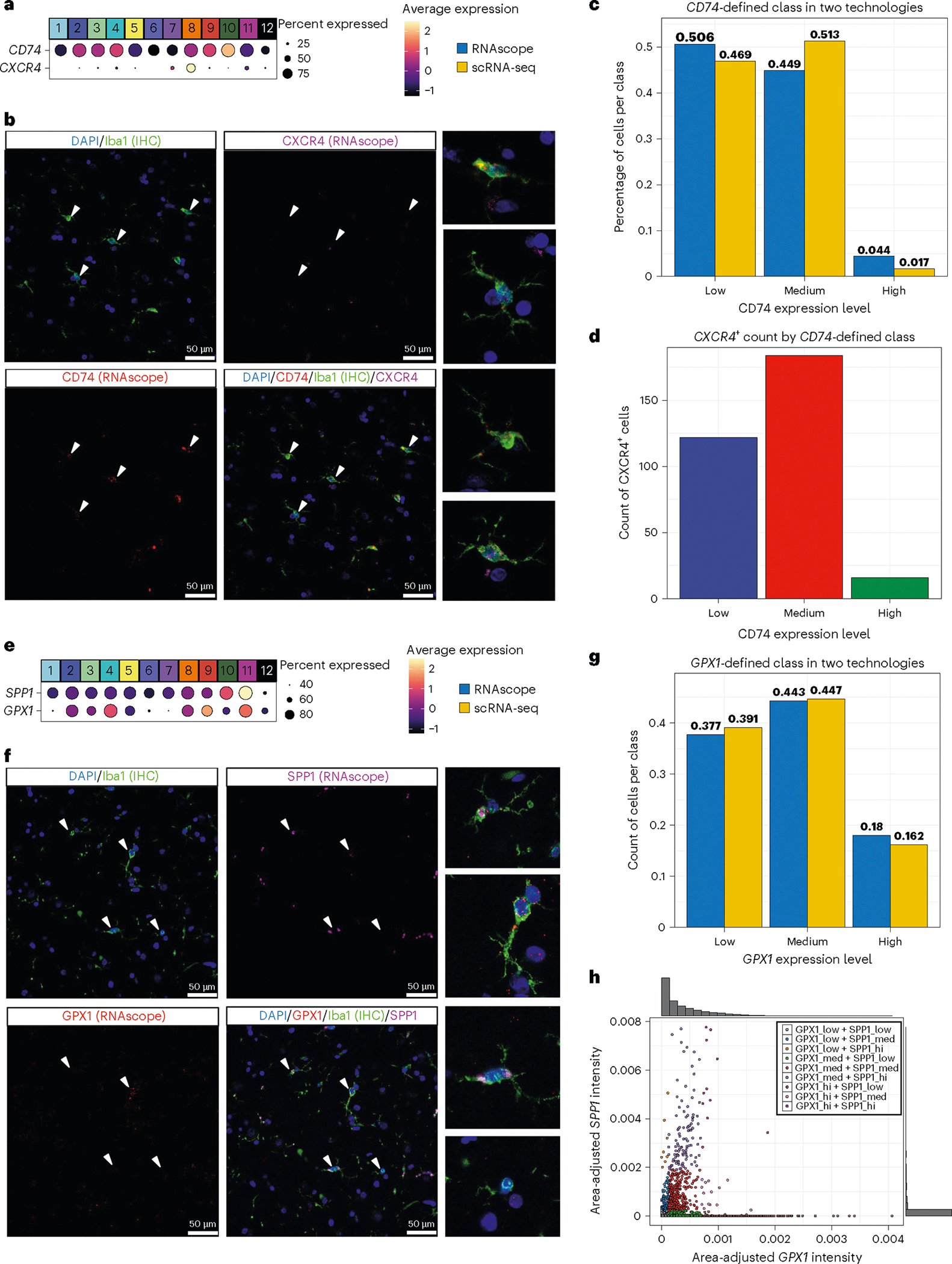
Human microglia play a pivotal role in neurological diseases, but we still have an incomplete understanding of microglial heterogeneity, which limits the development of targeted therapies directly modulating their state or function. Here, we use single-cell RNA sequencing to profile 215,680 live human microglia from 74 donors across diverse neurological diseases and CNS regions. We observe a central divide between oxidative and heterocyclic metabolism and identify microglial subsets associated with antigen presentation, motility and proliferation. Specific subsets are enriched in susceptibility genes for neurodegenerative diseases or the disease-associated microglial signature. We validate subtypes in situ with an RNAscope-immunofluorescence pipeline and high-dimensional MERFISH. We also leverage our dataset as a classification resource, finding that induced pluripotent stem cell model systems capture substantial in vivo heterogeneity. Finally, we identify and validate compounds that recapitulate certain subtypes in vitro, including camptothecin, which downregulates the signature of disease-enriched subtypes and upregulates a signature previously associated with Alzheimer's disease.
© 2024. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing interests: R.N.A. is funded by the NIH, DoD, the Parkinson’s Foundation and the Michael. J. Fox Foundation. R.N.A. received consultation fees from Avrobio, Caraway, GSK, Merck, Ono Therapeutics and Genzyme/Sanofi. P.L.D. has served as a consultant for Biogen, Merck-Serono and PureTech. All other authors declare no competing interests.
Figures














References
-
- Li Q & Barres BA Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 18, 225–242 (2018). - PubMed
MeSH terms
Grants and funding
- P30 AG072975/AG/NIA NIH HHS/United States
- U01 AG061356/AG/NIA NIH HHS/United States
- T32 GM007367/GM/NIGMS NIH HHS/United States
- P30 AG066462/AG/NIA NIH HHS/United States
- P30 AG010161/AG/NIA NIH HHS/United States
- U01 AG058589/AG/NIA NIH HHS/United States
- S10 OD020056/OD/NIH HHS/United States
- P30 CA013696/CA/NCI NIH HHS/United States
- RF1 AG057473/AG/NIA NIH HHS/United States
- U24 NS072026/NS/NINDS NIH HHS/United States
- R01 AG070438/AG/NIA NIH HHS/United States
- R01 AG015819/AG/NIA NIH HHS/United States
- F30 CA261090/CA/NCI NIH HHS/United States
- K01 AG084849/AG/NIA NIH HHS/United States
- U01 AG046152/AG/NIA NIH HHS/United States
- R01 AG017917/AG/NIA NIH HHS/United States
- P30 AG072980/AG/NIA NIH HHS/United States
- P30 AG019610/AG/NIA NIH HHS/United States
- R01 AG048015/AG/NIA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
