

Cardiovascular Disease May Be Triggered by Gut Microbiota, Microbial Metabolites, Gut Wall Reactions, and Inflammation
- PMID: 39408963
- PMCID: PMC11476619
- DOI: 10.3390/ijms251910634
Cardiovascular Disease May Be Triggered by Gut Microbiota, Microbial Metabolites, Gut Wall Reactions, and Inflammation
Abstract
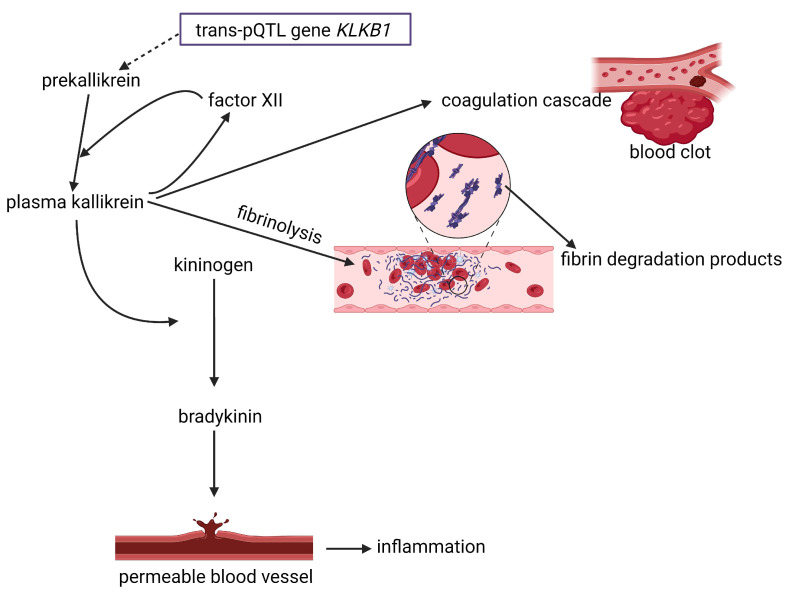
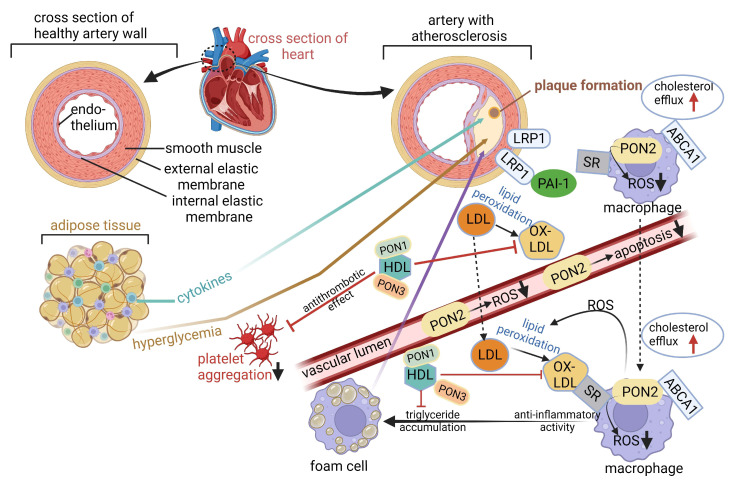
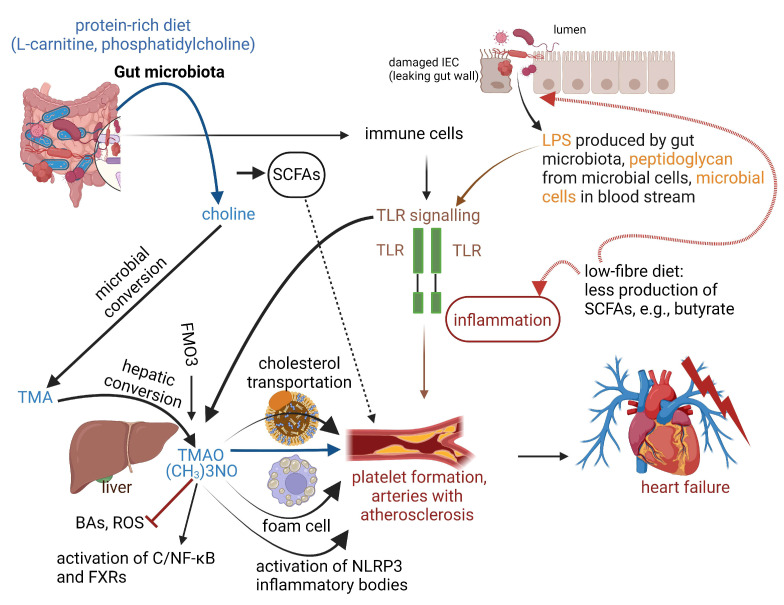
Cardiovascular disease (CVD) may be inherited, as recently shown with the identification of single nucleotide polymorphisms (SNPs or "snips") on a 250 kb DNA fragment that encodes 92 proteins associated with CVD. CVD is also triggered by microbial dysbiosis, microbial metabolites, metabolic disorders, and inflammatory intestinal epithelial cells (IECs). The epithelial cellular adhesion molecule (Ep-CAM) and trefoil factor 3 (TFF3) peptide keeps the gut wall intact and healthy. Variations in Ep-CAM levels are directly linked to changes in the gut microbiome. Leptin, plasminogen activator inhibitor 1 (PAI1), and alpha-1 acid glycoprotein 1 (AGP1) are associated with obesity and may be used as biomarkers. Although contactin 1 (CNTN1) is also associated with obesity and adiposity, it regulates the bacterial metabolism of tryptophan (Trp) and thus appetite. A decrease in CNTN1 may serve as an early warning of CVD. Short-chain fatty acids (SCFAs) produced by gut microbiota inhibit pro-inflammatory cytokines and damage vascular integrity. Trimethylamine N-oxide (TMAO), produced by gut microbiota, activates inflammatory Nod-like receptors (NLRs) such as Nod-like receptor protein 3 (NLRP3), which increase platelet formation. Mutations in the elastin gene (ELN) cause supra valvular aortic stenosis (SVAS), defined as the thickening of the arterial wall. Many of the genes expressed by human cells are regulated by gut microbiota. The identification of new molecular markers is crucial for the prevention of CVD and the development of new therapeutic strategies. This review summarizes the causes of CVD and identifies possible CVD markers.
Keywords: cardiovascular disease; gut microbiota; gut wall reactions; inflammation.
Conflict of interest statement
The author declares no conflicts of interest.
Figures
References
-
- Imhann F., Vila A.V., Bonder M.J., Fu J., Gevers D., Visschedijk M.C., Spekhorst L.M., Alberts R., Franke L., van Dullemen H.M., et al. Interplay of host genetics and gut microbiota underlying the onset and clinical presentation of inflammatory bowel disease. Gut. 2018;67:108–119. doi: 10.1136/gutjnl-2016-312135. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
