

Chemokine Receptor N-Terminus Charge Dictates Reliance on Post-Translational Modifications for Effective Ligand Capture and Following Boosting by Defense Peptides
- PMID: 39409188
- PMCID: PMC11477141
- DOI: 10.3390/ijms251910854
Chemokine Receptor N-Terminus Charge Dictates Reliance on Post-Translational Modifications for Effective Ligand Capture and Following Boosting by Defense Peptides
Abstract
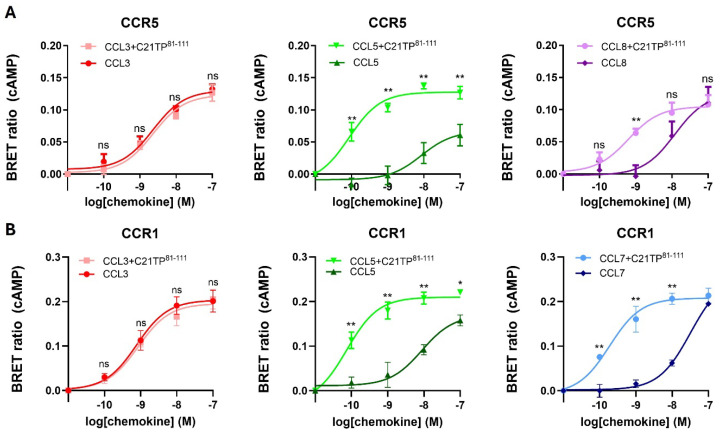
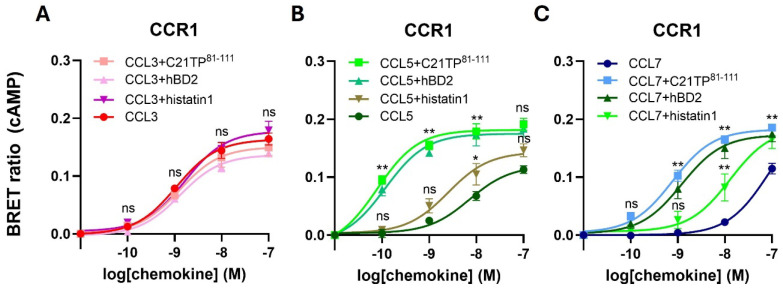
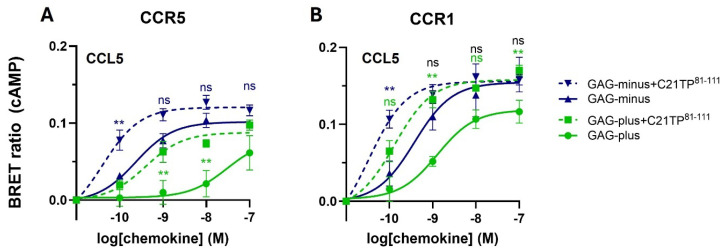
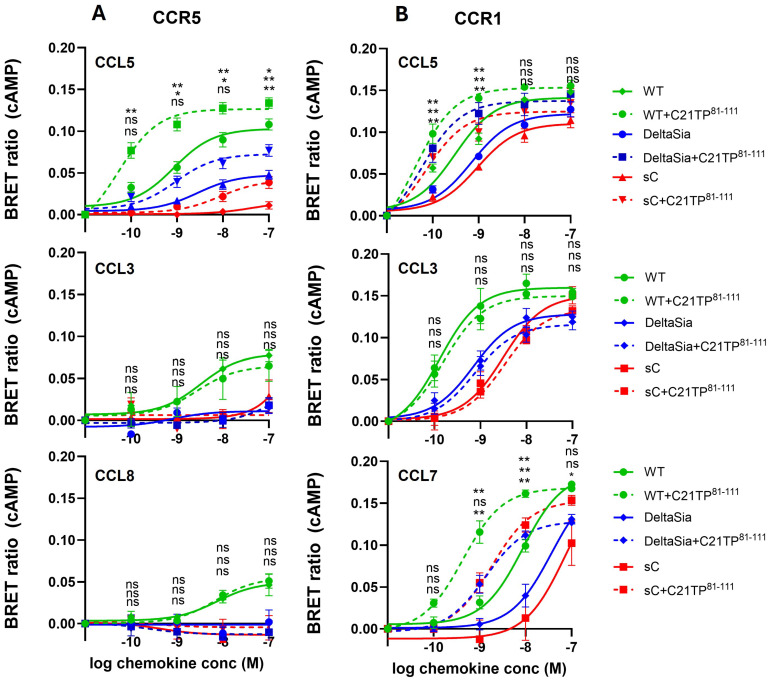
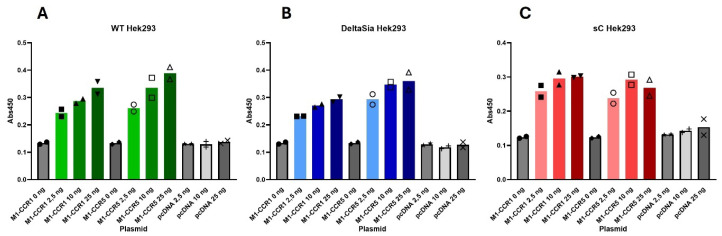
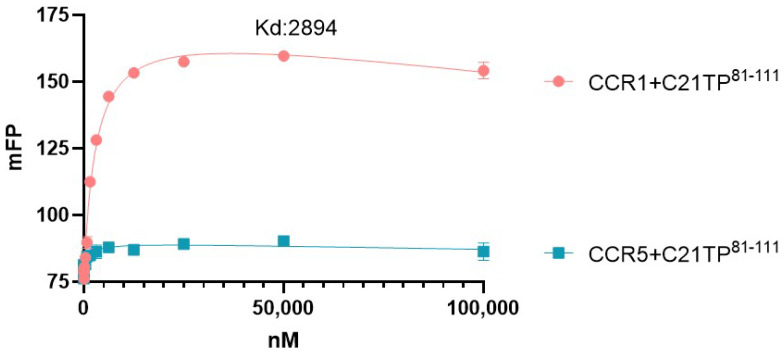
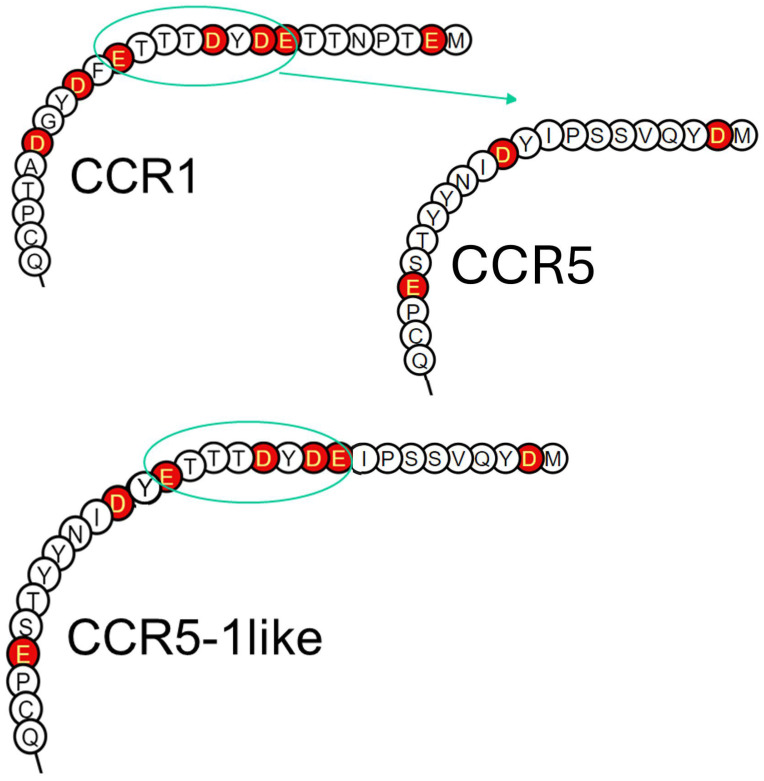
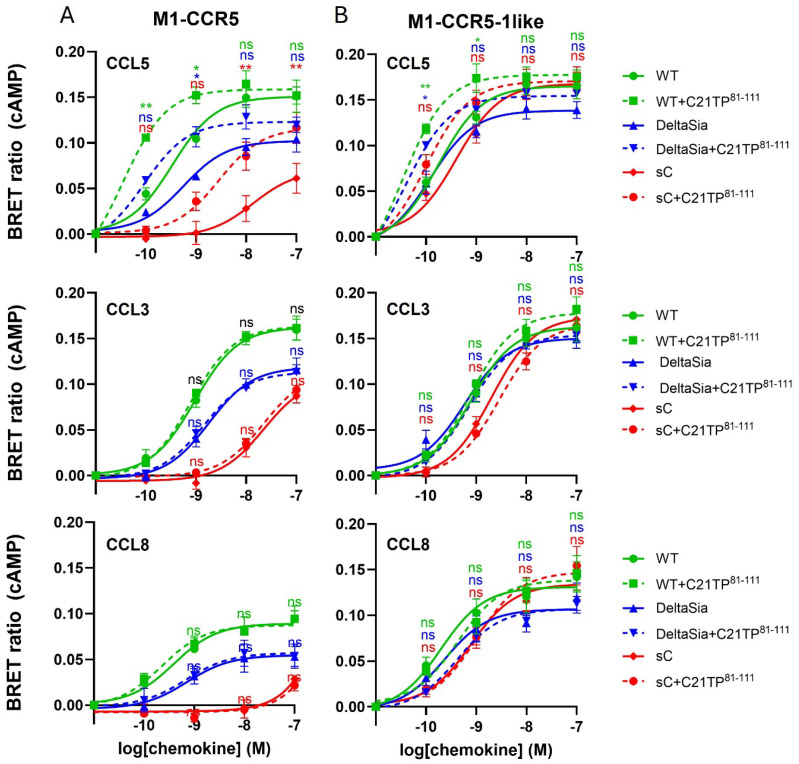
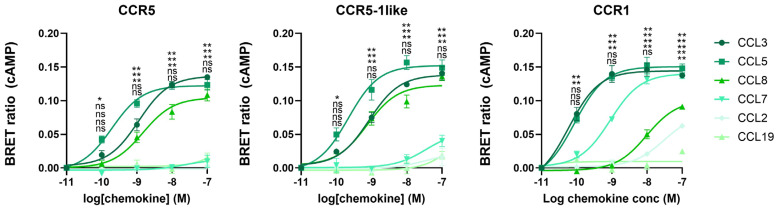
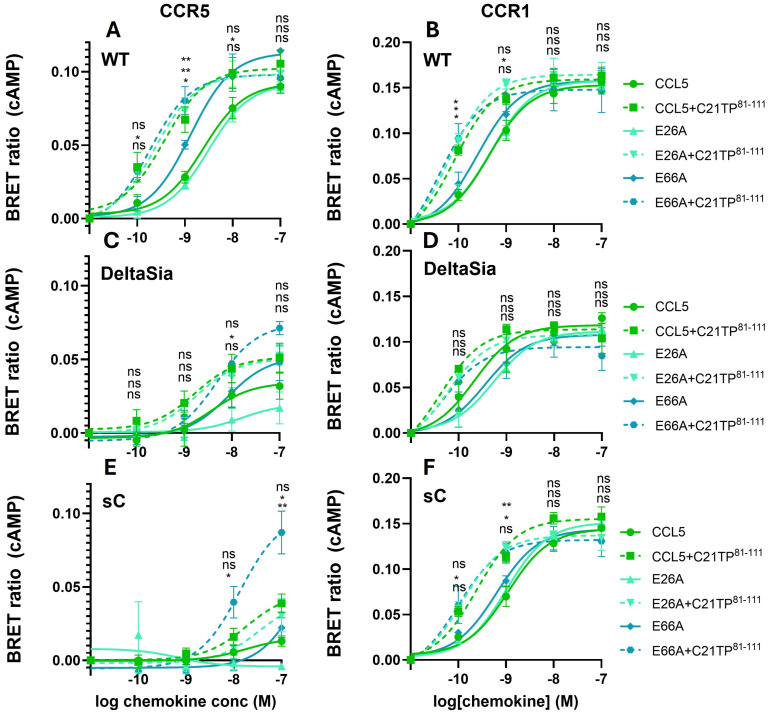
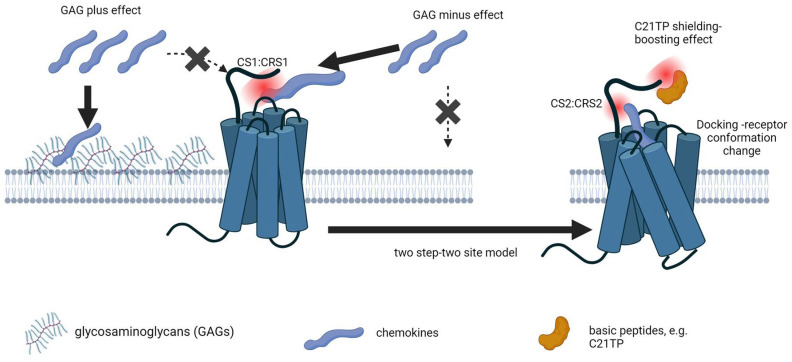
The chemokine receptors CCR1 and CCR5 display overlapping expression patterns and ligand dependency. Here we find that ligand activation of CCR5, not CCR1, is dependent on N-terminal receptor O-glycosylation. Release from O-glycosylation dependency is obtained by increasing CCR5 N-terminus acidity to the level of CCR1. Ligand activation of CCR5, not CCR1, drastically improves in the absence of glycosaminoglycans (GAGs). Ligand activity at both CCR1 and CCR5 is boosted by positively charged/basic peptides shown to interact with acidic chemokine receptor N-termini. We propose that receptors with an inherent low N-terminus acidity rely on post-translational modifications (PTMs) to efficiently compete with acidic entities in the local environment for ligand capture. Although crucial for initial ligand binding, strong electrostatic interactions between the ligand and the receptor N-terminus may counteract following insertion of the ligand into the receptor binding pocket and activation, a process that seems to be aided in the presence of basic peptides. Basic peptides bind to the naked CCR1 N-terminus, not the CCR5 N-terminus, explaining the loss of boosting of ligand-induced signaling via CCR5 in cells incapable of glycosylation.
Keywords: CCR1; CCR5; O-glycosylation; chemokine; electrostatic interaction; glycosaminoglycan; post-translational modification; receptor N-terminus; signaling; tyrosine sulfation.
Conflict of interest statement
B.F.V. and F.C.P. have ownership interests in Protein Foundry, LLC and XLock Bioscience, Inc. M.B.C. was employed by Evaxion Biotech and C.K.G. was employed by Glx Analytix APS. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as potential conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Figures
References
-
- Bachelerie F., Ben-Baruch A., Burkhardt A.M., Combadiere C., Farber J.M., Graham G.J., Horuk R., Sparre-Ulrich A.H., Locati M., Luster A.D., et al. International Union of Basic and Clinical Pharmacology. [corrected]. LXXXIX. Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacol. Rev. 2013;66:1–79. doi: 10.1124/pr.113.007724. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
