

snPATHO-seq, a versatile FFPE single-nucleus RNA sequencing method to unlock pathology archives
- PMID: 39414943
- PMCID: PMC11484811
- DOI: 10.1038/s42003-024-07043-2
snPATHO-seq, a versatile FFPE single-nucleus RNA sequencing method to unlock pathology archives
Abstract
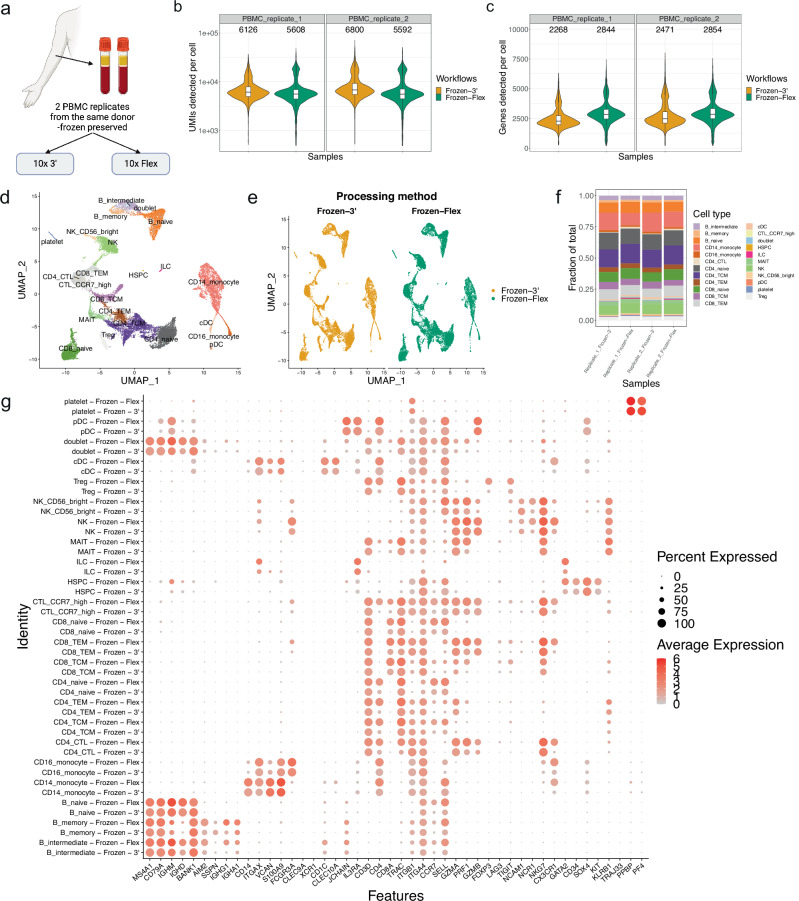
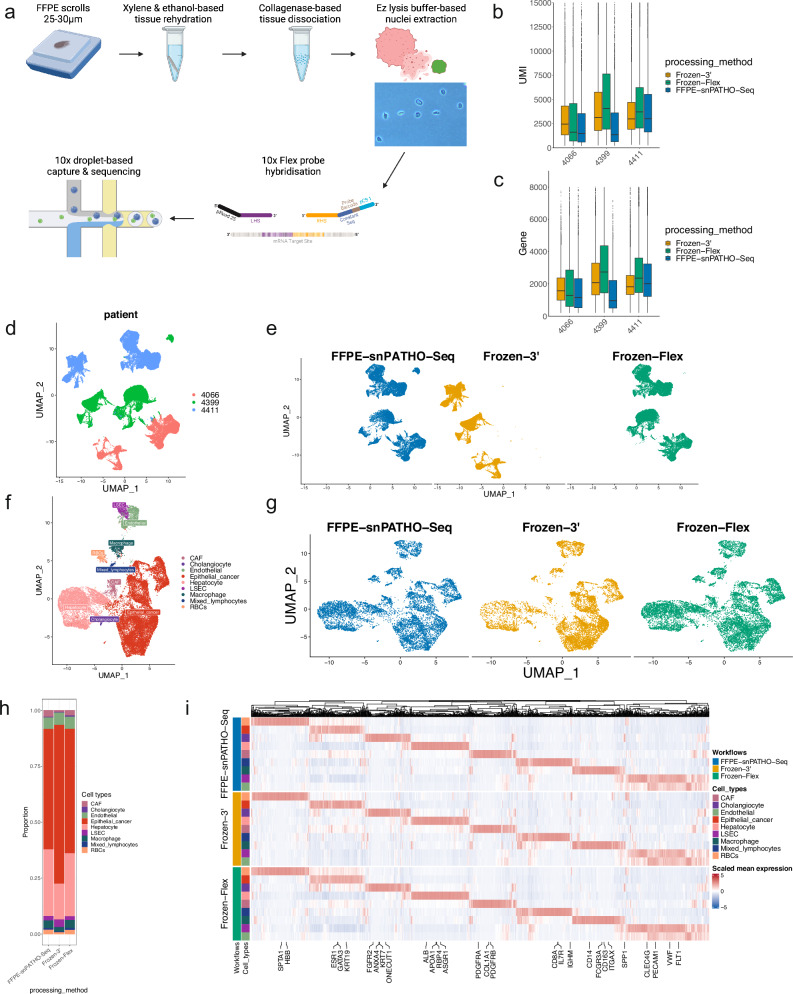
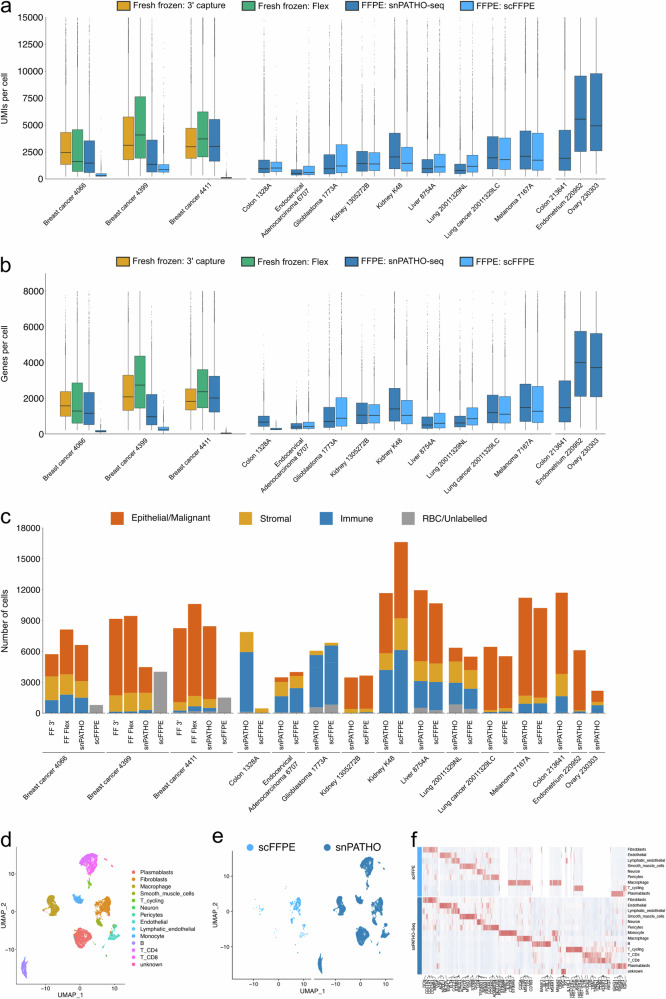
Formalin-fixed paraffin-embedded (FFPE) samples are valuable but underutilized in single-cell omics research due to their low RNA quality. In this study, leveraging a recent advance in single-cell genomic technology, we introduce snPATHO-seq, a versatile method to derive high-quality single-nucleus transcriptomic data from FFPE samples. We benchmarked the performance of the snPATHO-seq workflow against existing 10x 3' and Flex assays designed for frozen or fresh samples and highlighted the consistency in snRNA-seq data produced by all workflows. The snPATHO-seq workflow also demonstrated high robustness when tested across a wide range of healthy and diseased FFPE tissue samples. When combined with FFPE spatial transcriptomic technologies such as FFPE Visium, the snPATHO-seq provides a multi-modal sampling approach for FFPE samples, allowing more comprehensive transcriptomic characterization.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
