Knockdown of ketohexokinase versus inhibition of its kinase activity exert divergent effects on fructose metabolism
- PMID: 39418102
- PMCID: PMC11623947
- DOI: 10.1172/jci.insight.184396
Knockdown of ketohexokinase versus inhibition of its kinase activity exert divergent effects on fructose metabolism
Abstract
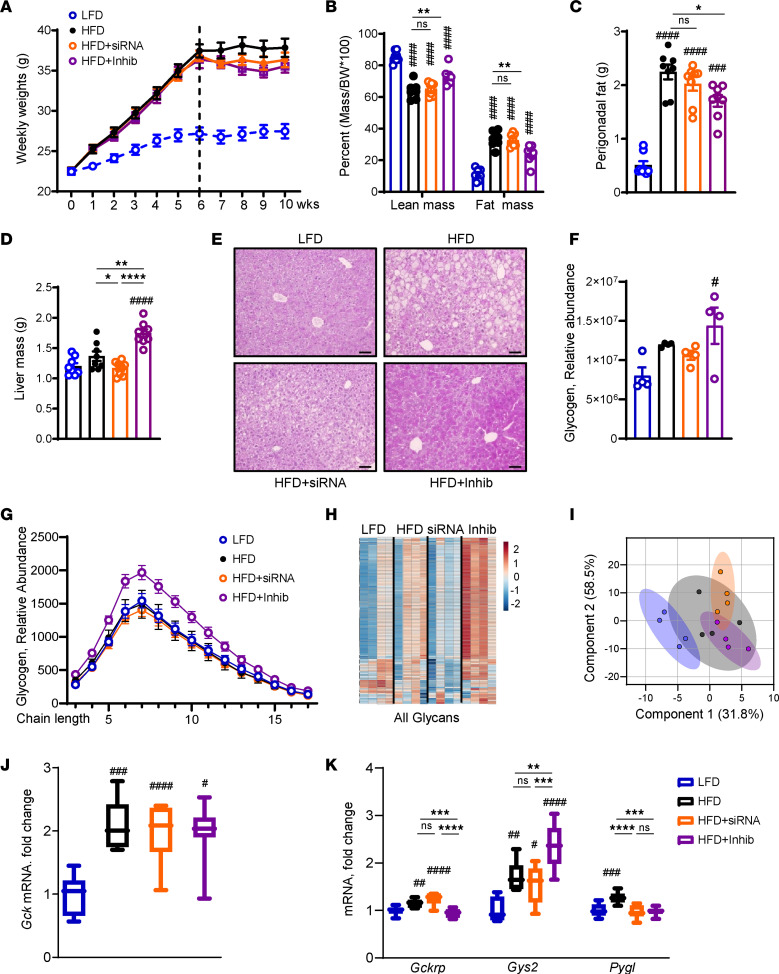
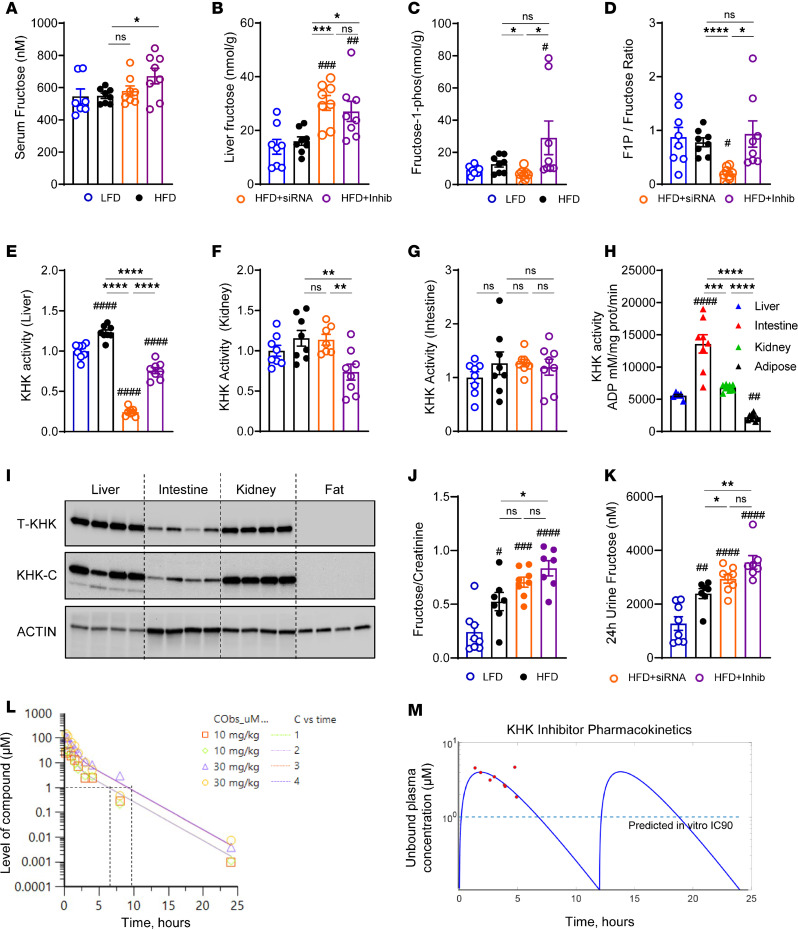
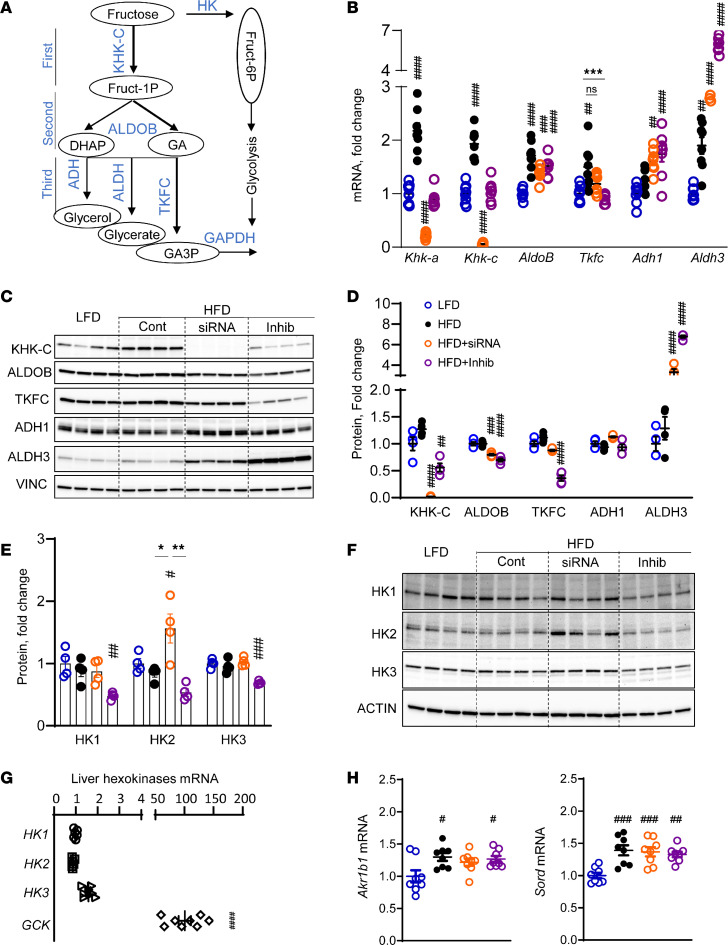
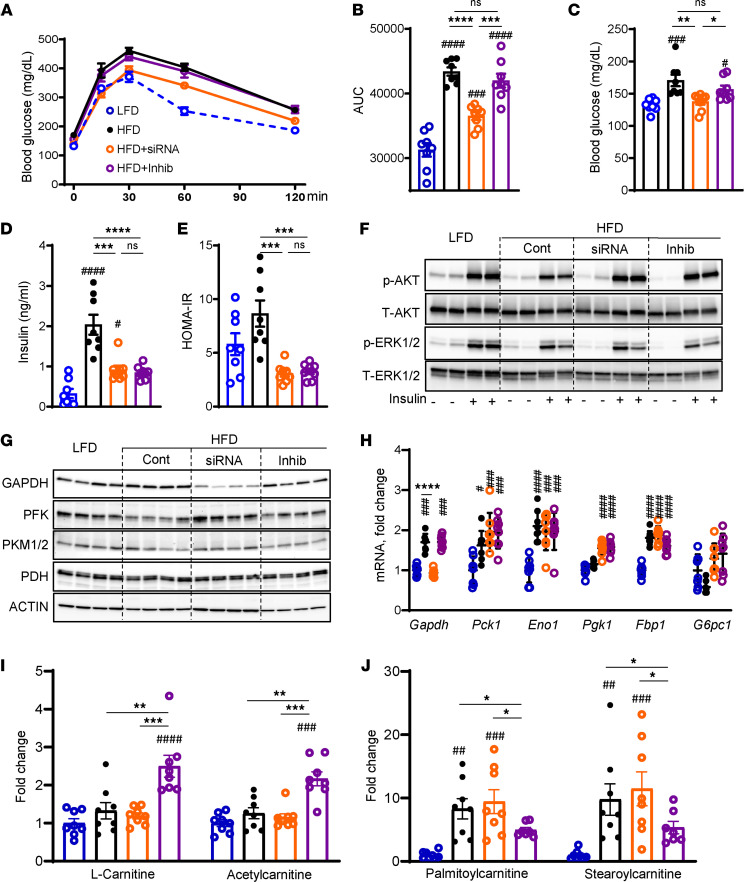
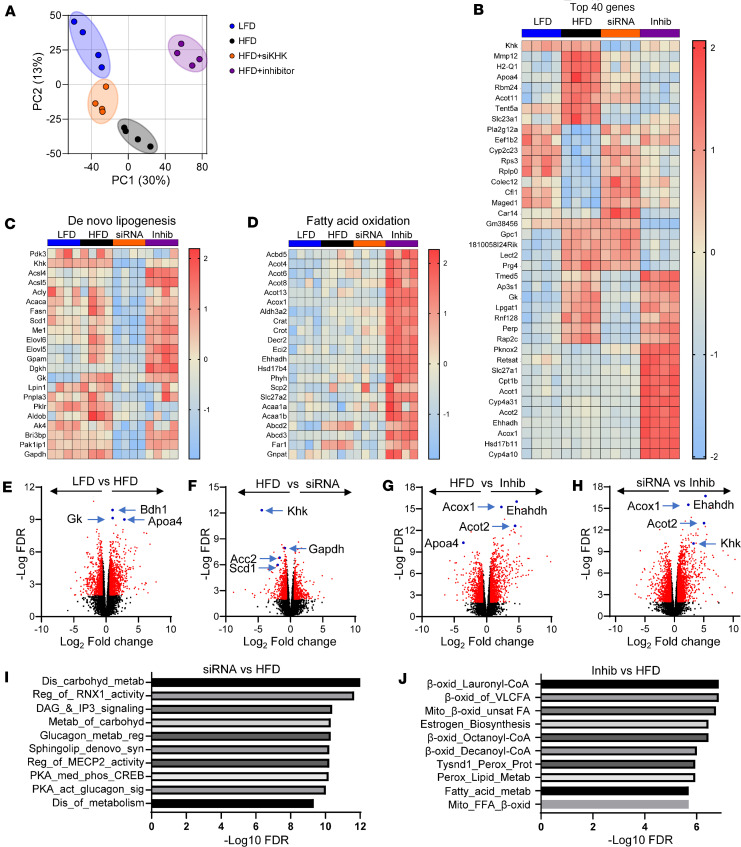
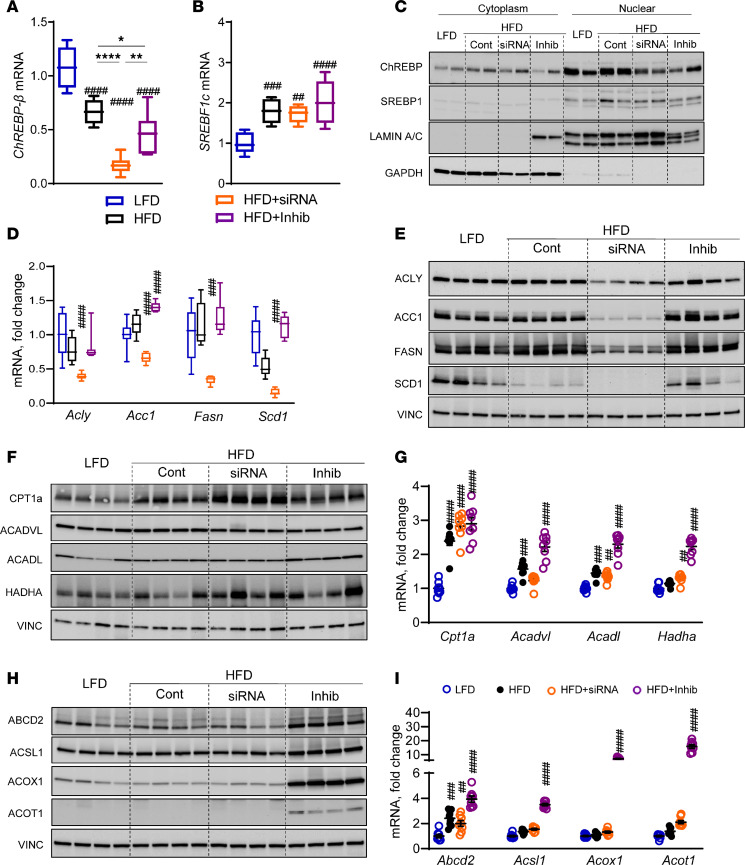
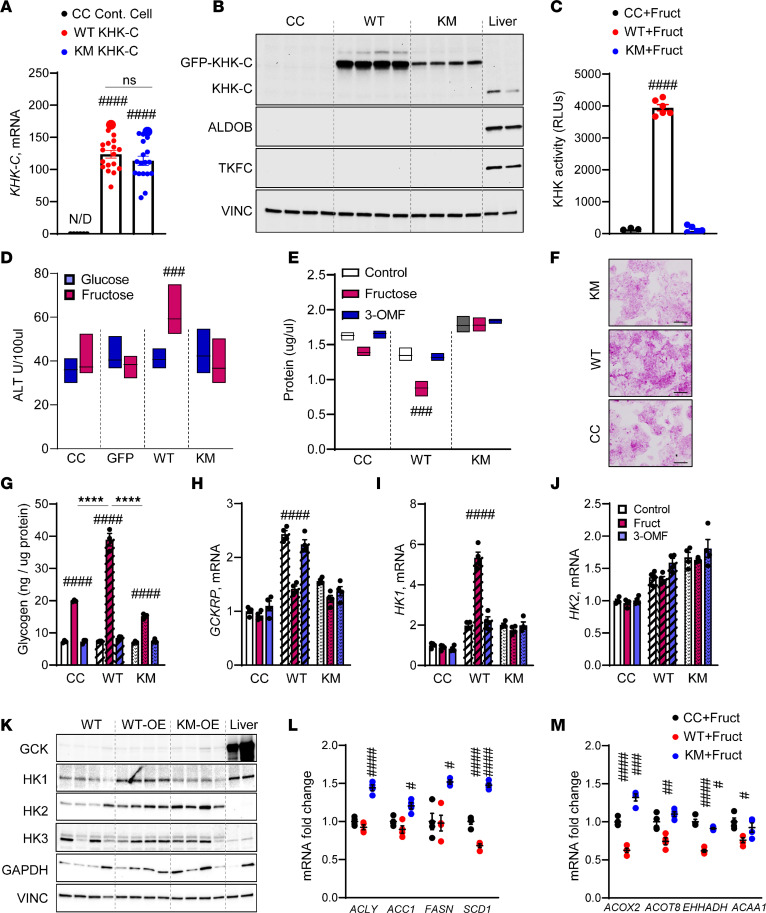
Excessive fructose intake is a risk factor for the development of obesity and its complications. Targeting ketohexokinase (KHK), the first enzyme of fructose metabolism, has been investigated for the management of metabolic dysfunction-associated steatotic liver disease (MASLD). We compared the effects of systemic, small molecule inhibitor of KHK enzymatic activity with hepatocyte-specific, N-acetylgalactosamine siRNA-mediated knockdown of KHK in mice on an HFD. We measured KHK enzymatic activity, extensively quantified glycogen accumulation, performed RNA-Seq analysis, and enumerated hepatic metabolites using mass spectrometry. Both KHK siRNA and KHK inhibitor led to an improvement in liver steatosis; however, via substantially different mechanisms, KHK knockdown decreased the de novo lipogenesis pathway, whereas the inhibitor increased the fatty acid oxidation pathway. Moreover, KHK knockdown completely prevented hepatic fructolysis and improved glucose tolerance. Conversely, the KHK inhibitor only partially reduced fructolysis, but it also targeted triokinase, mediating the third step of fructolysis. This led to the accumulation of fructose-1 phosphate, resulting in glycogen accumulation, hepatomegaly, and impaired glucose tolerance. Overexpression of wild-type, but not kinase-dead, KHK in cultured hepatocytes increased hepatocyte injury and glycogen accumulation after treatment with fructose. The differences between KHK inhibition and knockdown are, in part, explained by the kinase-dependent and -independent effects of KHK on hepatic metabolism.
Keywords: Carbohydrate metabolism; Hepatitis; Hepatology; Metabolism; Obesity.
Conflict of interest statement
Figures