

PP2A activation overcomes leptomeningeal dissemination in group 3 medulloblastoma
- PMID: 39419284
- PMCID: PMC11650719
- DOI: 10.1016/j.jbc.2024.107892
PP2A activation overcomes leptomeningeal dissemination in group 3 medulloblastoma
Abstract
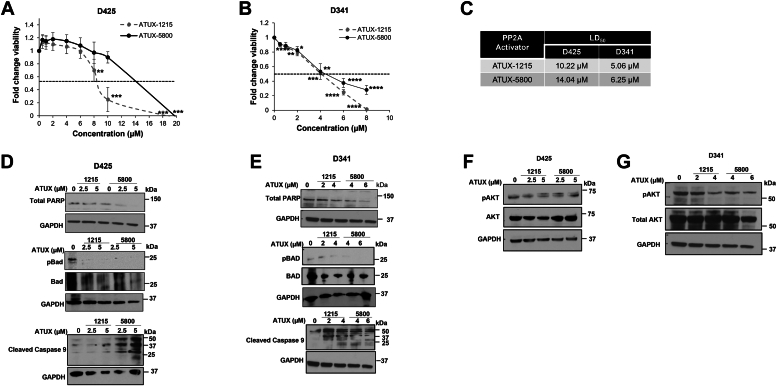
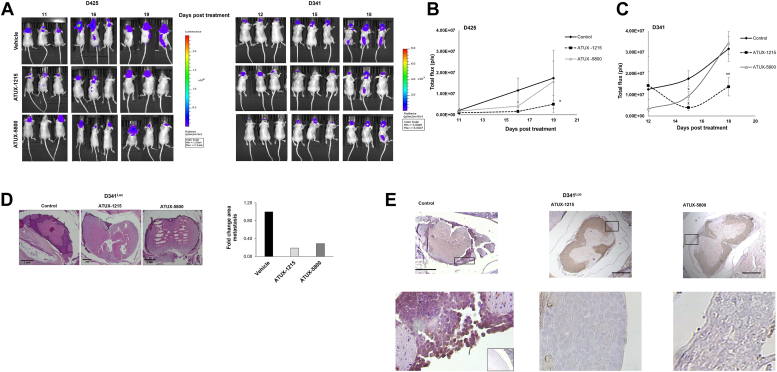
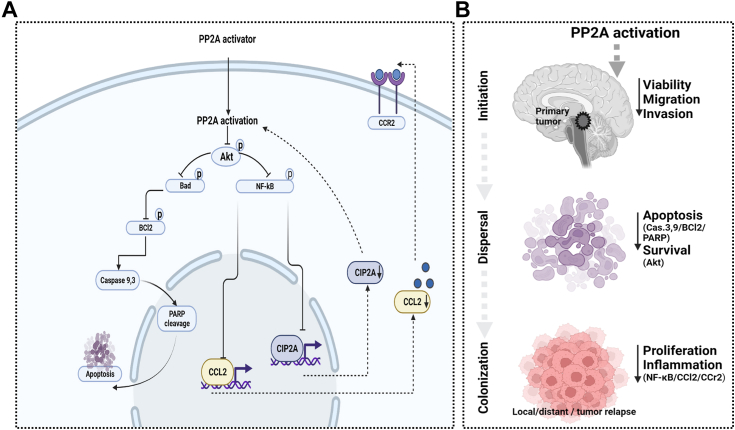
Leptomeningeal dissemination (LMD) is the primary cause of treatment failure in children with group 3 medulloblastoma (MB). Building on our previous work on protein phosphatase 2A (PP2A) activation in MB, here we present preclinical and molecular data on the effects of two novel classes of PP2A activators on disease processes of LMD in group 3 MB. The PP2A activators used in this study are ATUX-6156 and ATUX-6954 (diarylmethylcycloamine sulfonylureas), and ATUX-1215 and ATUX-5800 (diarylmethyl-4-aminotetrahydropyran-sulfonamides). Treatment with these compounds led to suppression of the endogenous PP2A inhibitor, cancerous inhibitor of PP2A (CIP2A), enhanced phosphatase activity (10-60%), and reduced MB viability, migration, and invasion, prerequisites for MB cells to access the cerebrospinal fluid, affecting the initiation stage of LMD. PP2A activator treatment of MB cells led to apoptosis mediated via caspase 9/PARP signaling due to decreased phosphorylation of Bad, impeding the dispersal stage of LMD. Cell proliferation and LMD-driving cellular traits and molecules pertinent to the third stage, colonization, were also affected. Treatment with ATUX-1215 or ATUX-5800 prevented LMD in an intraventricular murine model of MB, possibly mediated by disruption of the CCL2-CCR2 axis by altered NF-kB phosphorylation via disrupted AKT signaling. The present investigation offers proof-of-principle data for PP2A-based reactivation therapy for Group 3 MB and provides the first indications that PP2A reactivation may challenge the current paradigm in targeting the 3-stage process of MB LMD. Further investigations of PP2A activators are warranted as these compounds may prove beneficial as therapeutics for MB.
Keywords: PP2A; diarylmethyl cycloamine sulfonylureas; leptomeningeal dissemination; medulloblastoma; pyran sulfonamides.
Copyright © 2024 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interest Patent applications WO2023/023594 and WO2021/188949, inventor MO, are assigned to Atux Iskay LLC. The other authors declare that they have no conflict of interest with the contents of this article.
Figures





References
-
- Sung H., Ferlay J., Siegel R.L., Laversanne M., Soerjomataram I., Jemal A., et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality Worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021;71:209–249. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
