

The integrated molecular and histological analysis defines subtypes of esophageal squamous cell carcinoma
- PMID: 39419971
- PMCID: PMC11487165
- DOI: 10.1038/s41467-024-53164-x
The integrated molecular and histological analysis defines subtypes of esophageal squamous cell carcinoma
Abstract
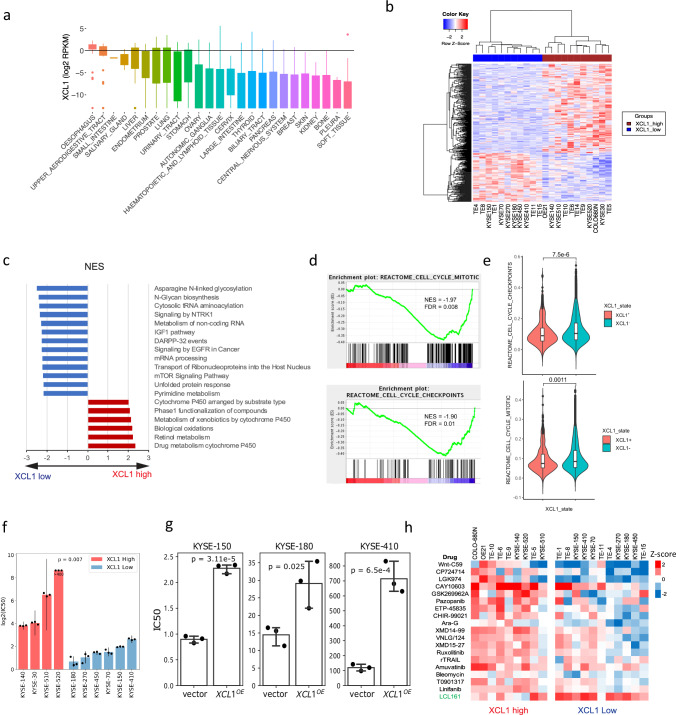
Esophageal squamous cell carcinoma (ESCC) is highly heterogeneous. Our understanding of full molecular and immune landscape of ESCC remains limited, hindering the development of personalised therapeutic strategies. To address this, we perform genomic-transcriptomic characterizations and AI-aided histopathological image analysis of 120 Chinese ESCC patients. Here we show that ESCC can be categorized into differentiated, metabolic, immunogenic and stemness subtypes based on bulk and single-cell RNA-seq, each exhibiting specific molecular and histopathological features based on an amalgamated deep-learning model. The stemness subgroup with signature genes, such as WFDC2, SFRP1, LGR6 and VWA2, has the poorest prognosis and is associated with downregulated immune activities, a high frequency of EP300 mutation/activation, functional mutation enrichment in Wnt signalling and the highest level of intratumoural heterogeneity. The immune profiling by transcriptomics and immunohistochemistry reveals ESCC cells overexpress natural killer cell markers XCL1 and CD160 as immune evasion. Strikingly, XCL1 expression also affects the sensitivity of ESCC cells to common chemotherapy drugs. This study opens avenues for ESCC treatment and provides a valuable public resource to better understand ESCC.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures




References
Publication types
MeSH terms
Substances
Associated data
- BioProject/PRJCA001577
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
