

A systematic review and meta-analysis of GFAP gene variants in Alexander disease
- PMID: 39420046
- PMCID: PMC11487261
- DOI: 10.1038/s41598-024-75383-4
A systematic review and meta-analysis of GFAP gene variants in Alexander disease
Abstract
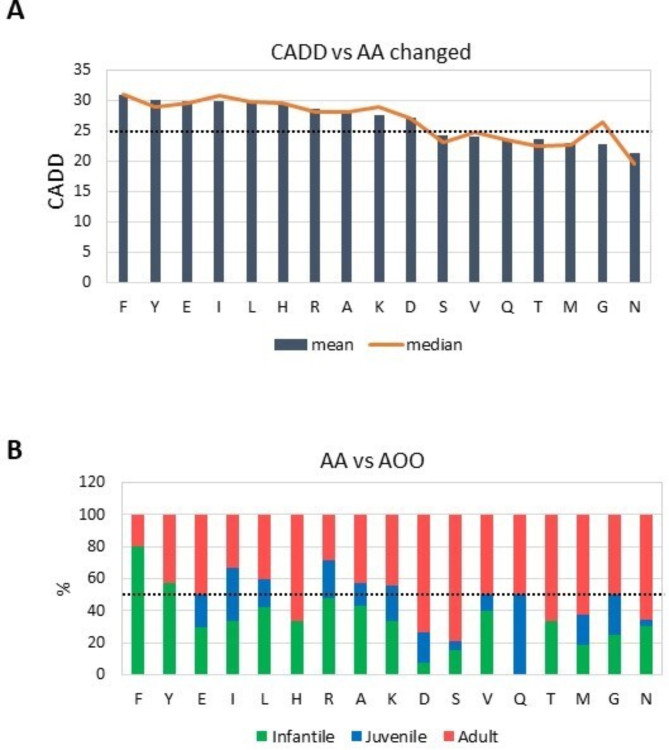
Alexander disease (ALXDRD) is a rare neurodegenerative disorder of astrocytes resulting from pathogenic variants in the GFAP gene. The genotype-phenotype correlation remains elusive due to the variable expressivity of clinical manifestations. In an attempt to clarify the effects of GFAP variants in ALXDRD, numerous studies were collected and analyzed. In particular, we systematically searched for GFAP variants associated with ALXDRD and collected information on the location within the gene and protein, prediction of deleteriousness/pathogenicity, occurrence, sex and country of origin of patients, DNA source, genetic testing, and clinical signs. To identify possible associations, statistical analyses and meta-analyses were applied, thus revealing a higher than expected percentage of adult patients with ALXDRD. Furthermore, substitution of Arginine, the most frequently altered residue among the 550 predominantly missense causative GFAP variants collected, were mostly de novo and more prevalent in early-onset forms of ALXDRD. The effect of defective splicing in modifying the impact of GFAP variants on the age of onset of ALXDRD was also postulated after evaluating the distribution of the corresponding deleterious predictive values. In conclusion, not only previously unrecognized genotype-phenotype correlations were revealed in ALXDRD, but also subtle mechanisms could explain the variable manifestations of the ALXDRD clinical phenotype.
Keywords: Alexander Disease; GFAP; Genotype-phenotype correlation; Meta-analysis; Variant effect.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures



References
-
- Russo, L. S., Aron, A. & Anderson, P. J. Alexander’s disease: a report and reappraisal. Neurol. 26, 607–614 (1976). - PubMed
-
- Springer, S. et al. Alexander disease–classification revisited and isolation of a neonatal form. Neuropediatrics. 31, 86–92 (2000). - PubMed
-
- Yoshida, T. et al. Nationwide survey of Alexander disease in Japan and proposed new guidelines for diagnosis. J. Neurol. 258, 1998–2008 (2011). - PubMed
-
- Nielsen, A. L., Jørgensen, P. & Jørgensen, A. L. Mutations associated with a childhood leukodystrophy, Alexander disease, cause deficiency in dimerization of the cytoskeletal protein GFAP. J. Neurogenet. 16, 175–179 (2002). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
