

Genetic architecture of oral glucose-stimulated insulin release provides biological insights into type 2 diabetes aetiology
- PMID: 39420167
- PMCID: PMC11496110
- DOI: 10.1038/s42255-024-01140-6
Genetic architecture of oral glucose-stimulated insulin release provides biological insights into type 2 diabetes aetiology
Abstract
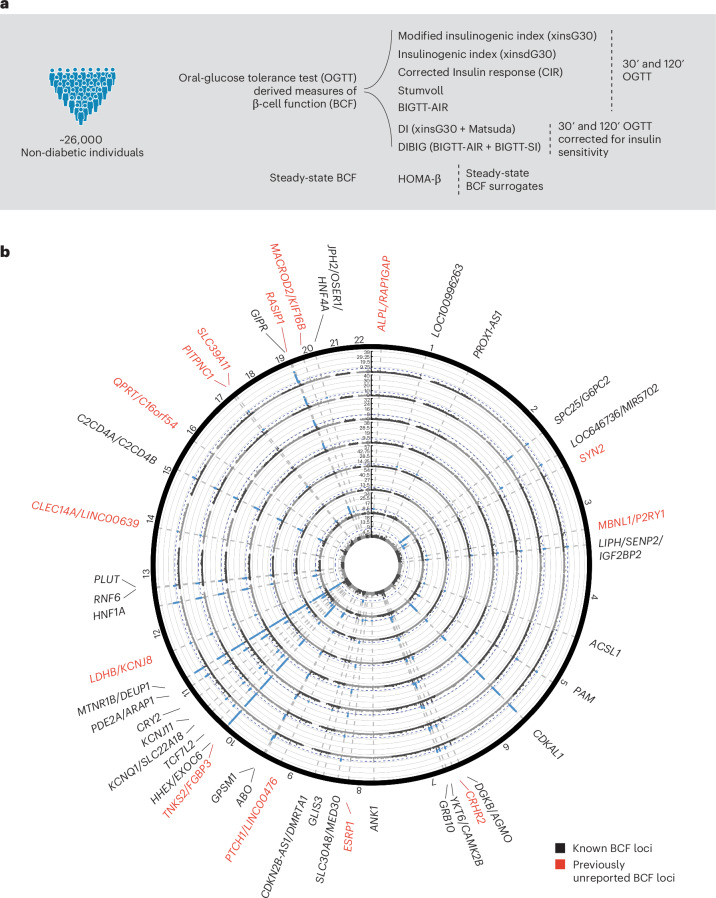
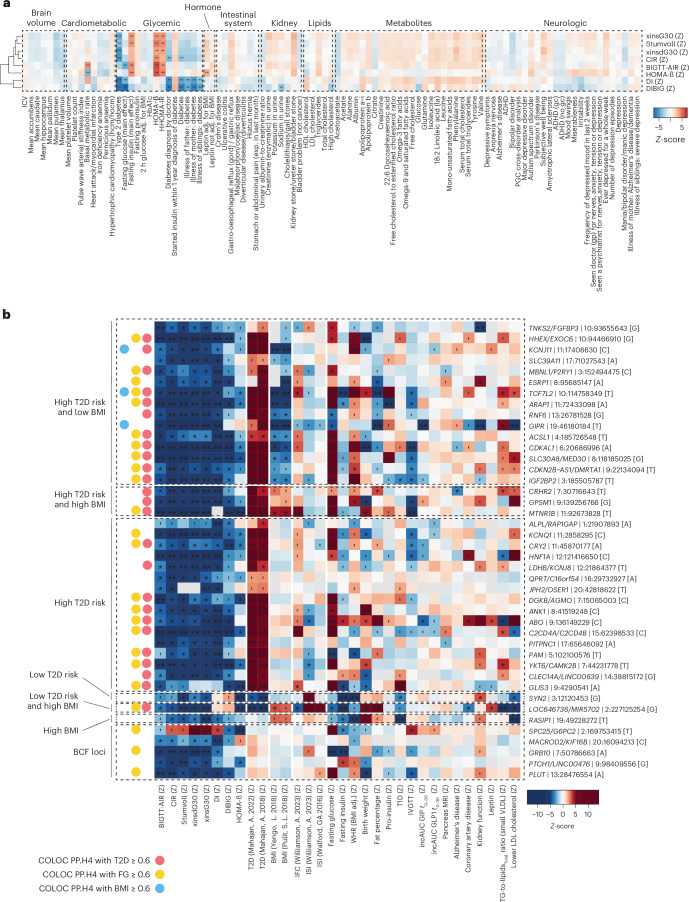
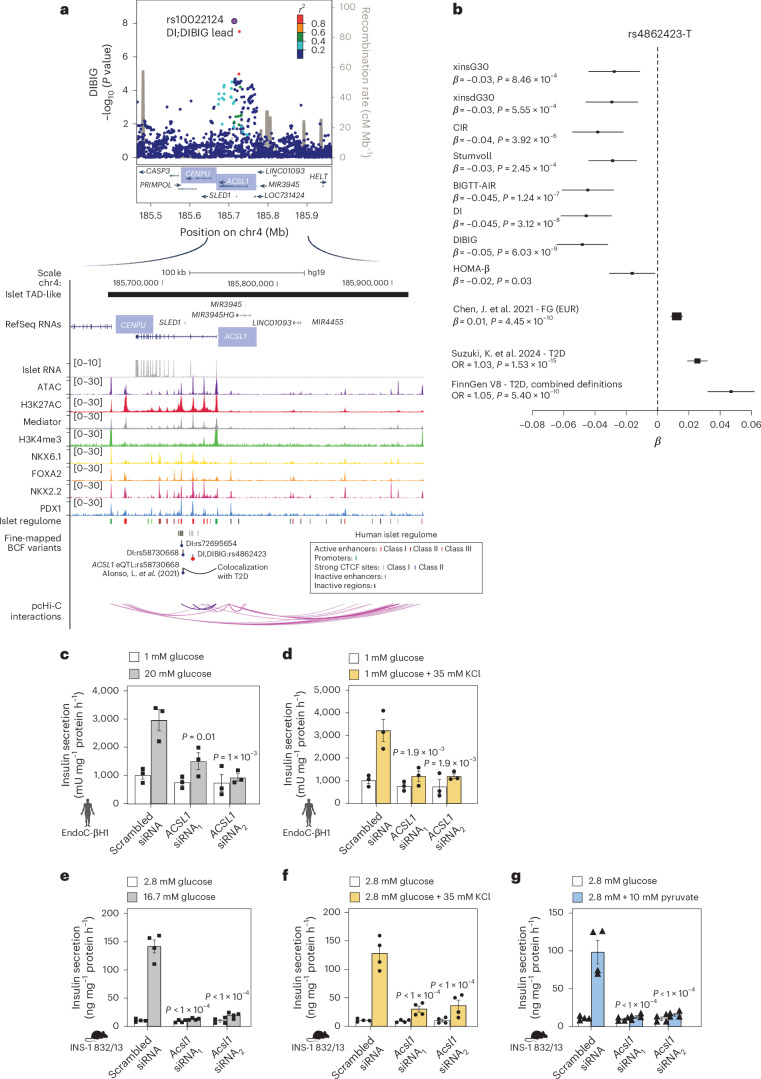
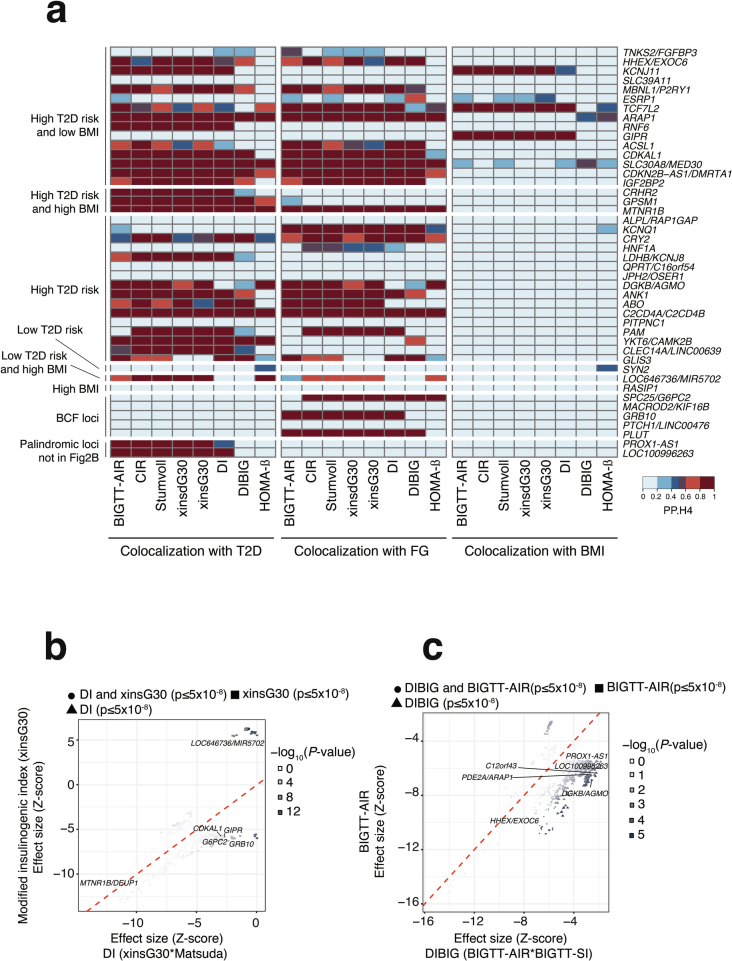
The genetics of β-cell function (BCF) offer valuable insights into the aetiology of type 2 diabetes (T2D)1,2. Previous studies have expanded the catalogue of BCF genetic associations through candidate gene studies3-7, large-scale genome-wide association studies (GWAS) of fasting BCF8,9 or functional islet studies on T2D risk variants10-14. Nonetheless, GWAS focused on BCF traits derived from oral glucose tolerance test (OGTT) data have been limited in sample size15,16 and have often overlooked the potential for related traits to capture distinct genetic features of insulin-producing β-cells17,18. We reasoned that investigating the genetic basis of multiple BCF estimates could provide a broader understanding of β-cell physiology. Here, we aggregate GWAS data of eight OGTT-based BCF traits from ~26,000 individuals of European descent, identifying 55 independent genetic associations at 44 loci. By examining the effects of BCF genetic signals on related phenotypes, we uncover diverse disease mechanisms whereby genetic regulation of BCF may influence T2D risk. Integrating BCF-GWAS data with pancreatic islet transcriptomic and epigenomic datasets reveals 92 candidate effector genes. Gene silencing in β-cell models highlights ACSL1 and FAM46C as key regulators of insulin secretion. Overall, our findings yield insights into the biology of insulin release and the molecular processes linking BCF to T2D risk, shedding light on the heterogeneity of T2D pathophysiology.
© 2024. The Author(s).
Conflict of interest statement
G.A. became a full-time employee of Novo Nordisk Ltd. and N.G. became a full-time employee at Novo Nordisk A/S while this manuscript was being drafted. The remaining authors declare no competing interests.
Figures










References
-
- Bonnefond, A. & Froguel, P. Rare and common genetic events in type 2 diabetes: What should biologists know? Cell Metab.21, 357–368 (2015). - PubMed
-
- Rose, C. S. et al. A −30G>A polymorphism of the β-cell-specific glucokinase promoter associates with hyperglycemia in the general population of whites. Diabetes54, 3026–3031 (2005). - PubMed
-
- Grarup, N. et al. Studies of association of variants near the HHEX, CDKN2A/B, and IGF2BP2 genes with type 2 diabetes and impaired insulin release in 10,705 Danish subjects. Diabetes56, 3105–3111 (2007). - PubMed
MeSH terms
Substances
Grants and funding
- R01 DK093757/DK/NIDDK NIH HHS/United States
- R56 DK062370/DK/NIDDK NIH HHS/United States
- R01 DK072193/DK/NIDDK NIH HHS/United States
- WT101033/Wellcome Trust (Wellcome)
- R01 DK062370/DK/NIDDK NIH HHS/United States
- PMP21/00067/Ministry of Economy and Competitiveness | Instituto de Salud Carlos III (Institute of Health Carlos III)
- ZIA HG000024/ImNIH/Intramural NIH HHS/United States
- NNF18OC0034532/Novo Nordisk Fonden (Novo Nordisk Foundation)
- U01 DK062370/DK/NIDDK NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
