

Slowly progressive autosomal dominant Alport Syndrome due to COL4A3 splicing variant
- PMID: 39424670
- PMCID: PMC11985956
- DOI: 10.1038/s41431-024-01706-8
Slowly progressive autosomal dominant Alport Syndrome due to COL4A3 splicing variant
Erratum in
-
Correction: Slowly progressive autosomal dominant Alport Syndrome due to COL4A3 splicing variant.Eur J Hum Genet. 2025 Apr;33(4):556-557. doi: 10.1038/s41431-024-01736-2. Eur J Hum Genet. 2025. PMID: 39587357 Free PMC article. No abstract available.
Abstract
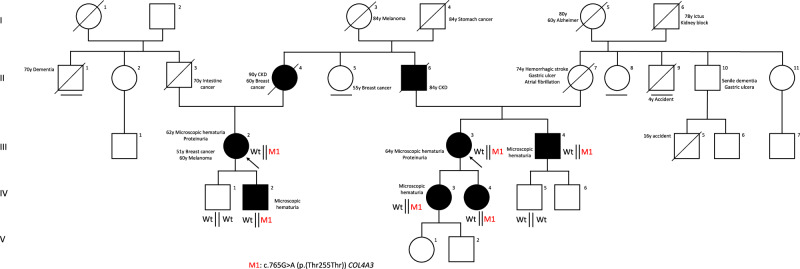
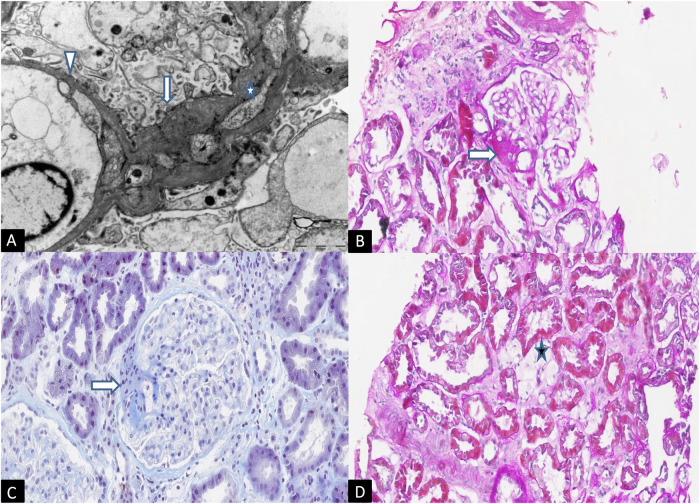
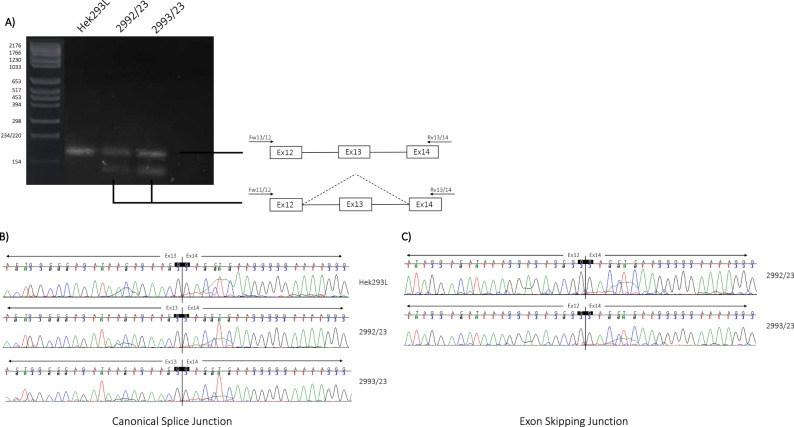
Alport syndrome is a rare genetic kidney disease caused by variants in the COL4A3/A4/A5 genes. It's characterised by progressive kidney failure, though therapies targeting Renin-Angiotensin System can delay its progression. Additionally, extrarenal manifestations may sometimes coexist. Recent advances in genetic analysis and the necessity to better clarify genotype-phenotype correlations in affected patients raises the importance of detecting even cryptic splicing variants, lying in both canonical and non-canonical splice sites variants such as last exonic nucleotide variants. These variants, often, do not cause an amino acid change but alter the snRNP proteins binding. We studied a big Italian family with Alport syndrome showing a clear dominant pattern of transmission with younger family members having only haematuria and older individuals presenting with End-Stage Kidney Failure (ESKF). Kidney biopsy showed the typical disease hallmarks. We deeply mined the data for SNV and CNV through exome sequencing on DNA from both peripheral blood samples and patients' podocytes-lineage cells. We identified an already reported synonymous variant, c.765G>A (p.(Thr255Thr)), in the last exonic nucleotide of exon 13 of the COL4A3 gene. Employing the patient's podocytes we demonstrated that this variant results in exon skipping leading to an in-frame deletion of 28 amino acids without leaky effect. According to the pattern of transmission, to the kidney biopsy and to the exome data analysis we provided further evidence that autosomal dominant Alport syndrome is a well-defined clinical entity. We also confirmed the pathogenicity of the synonymous COL4A3 variant for the first time demonstrating its role in a dominant pattern of transmission.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests. Ethical approval: The study was approved by Azienda Ospedaliera Universitaria Senese Ethics Commitee, Prot Name IPSA, Prot n 12030_2017 on 12.18.2017. Informed consent: Informed consent was provided.
Figures

References
-
- Jais JP, Knebelmann B, Giatras I, Marchi M, Rizzoni G, Renieri A, et al. X-linked Alport syndrome: natural history in 195 families and genotype- phenotype correlations in males. J Am Soc Nephrol. 2000;11:649–57. - PubMed
-
- Jais JP, Knebelmann B, Giatras I, De Marchi M, Rizzoni G, Renieri A, et al. X-linked Alport syndrome: natural history and genotype-phenotype correlations in girls and women belonging to 195 families: a “European Community Alport Syndrome Concerted Action” study. J Am Soc Nephrol. 2003;14:2603–10. - PubMed
-
- Longo I, Porcedda P, Mari F, Giachino D, Meloni I, Deplano C, et al. COL4A3/COL4A4 mutations: from familial haematuria to autosomal-dominant or recessive Alport syndrome. Kidney Int. 2002;61:1947–56. - PubMed
-
- Longo I, Scala E, Mari F, Caselli R, Pescucci C, Mencarelli MA, et al. Autosomal recessive Alport syndrome: an in-depth clinical and molecular analysis of five families. Nephrol Dial Transpl. 2006;21:665–71. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
