

Droplet Hi-C enables scalable, single-cell profiling of chromatin architecture in heterogeneous tissues
- PMID: 39424717
- PMCID: PMC12520981
- DOI: 10.1038/s41587-024-02447-1
Droplet Hi-C enables scalable, single-cell profiling of chromatin architecture in heterogeneous tissues
Erratum in
-
Author Correction: Droplet Hi-C enables scalable, single-cell profiling of chromatin architecture in heterogeneous tissues.Nat Biotechnol. 2025 Oct;43(10):1728. doi: 10.1038/s41587-025-02697-7. Nat Biotechnol. 2025. PMID: 40355567 Free PMC article. No abstract available.
Abstract
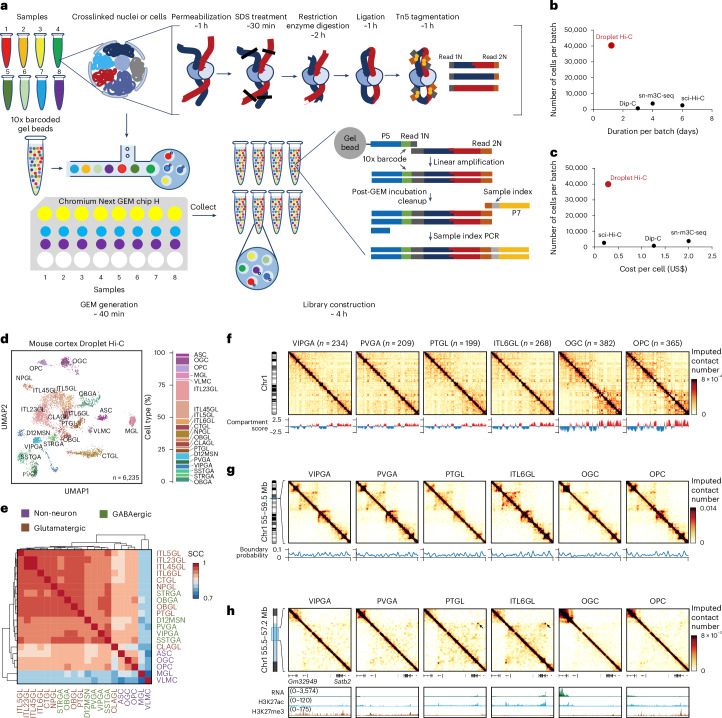
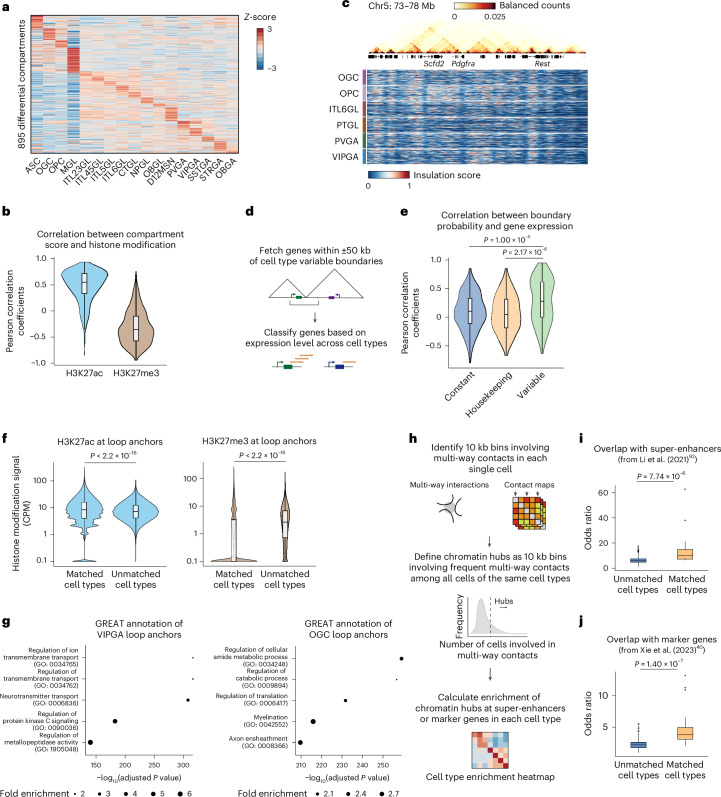
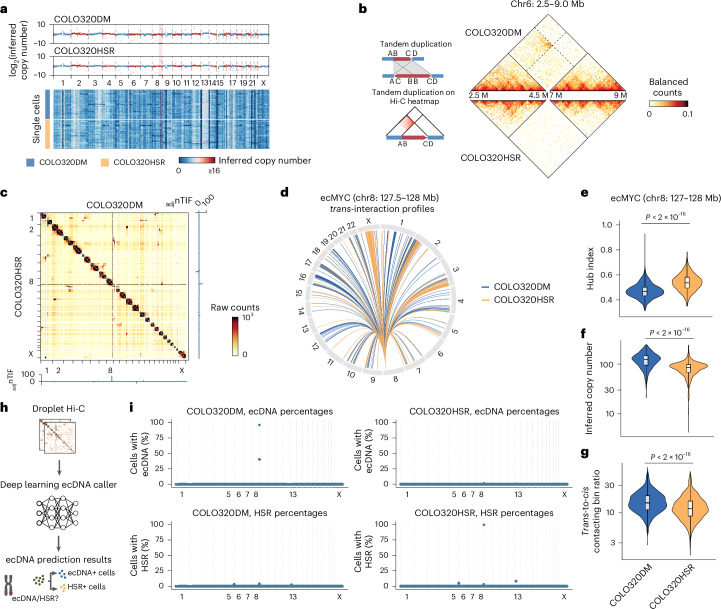
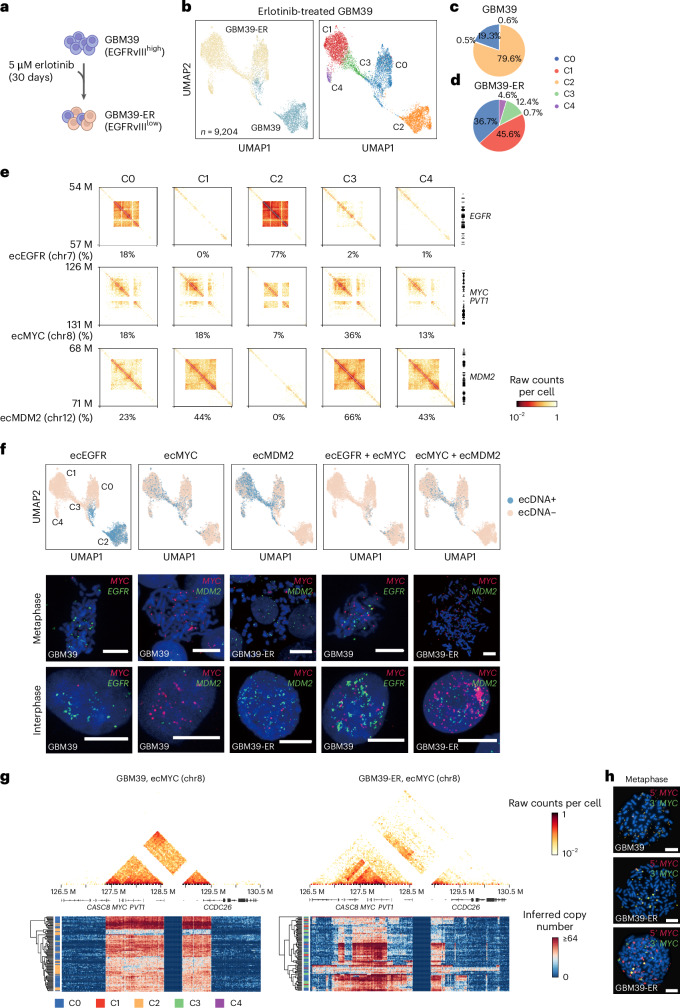
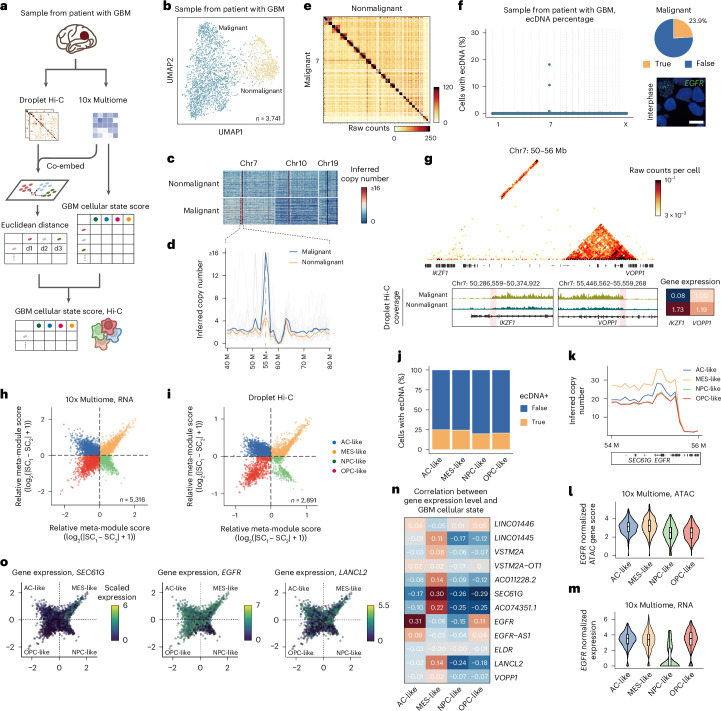
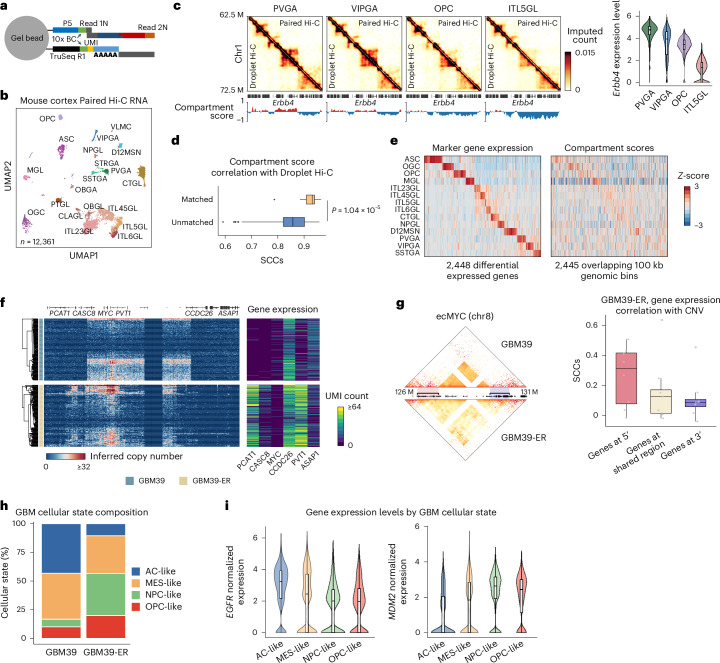
Current methods for analyzing chromatin architecture are not readily scalable to heterogeneous tissues. Here we introduce Droplet Hi-C, which uses a commercial microfluidic device for high-throughput, single-cell chromatin conformation profiling in droplets. Using Droplet Hi-C, we mapped the chromatin architecture of the mouse cortex and analyzed gene regulatory programs in major cortical cell types. In addition, we used this technique to detect copy number variations, structural variations and extrachromosomal DNA in human glioblastoma, colorectal and blood cancer cells, revealing clonal dynamics and other oncogenic events during treatment. We refined the technique to allow joint profiling of chromatin architecture and transcriptome in single cells, facilitating exploration of the links between chromatin architecture and gene expression in both normal tissues and tumors. Thus, Droplet Hi-C both addresses critical gaps in chromatin analysis of heterogeneous tissues and enhances understanding of gene regulation.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: B.R. is a cofounder of Epigenome Technologies, Inc. and has equity in Arima Genomics, Inc. The other authors declare no competing interests.
Figures










References
MeSH terms
Substances
Grants and funding
- 1UM1HG011585/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- R35HG011922/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- R01 CA258248/CA/NCI NIH HHS/United States
- P30 NS047101/NS/NINDS NIH HHS/United States
- 5T32GM007198-47/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- 5T32CA067754-27/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- S10 OD026929/OD/NIH HHS/United States
- UM1 HG011585/HG/NHGRI NIH HHS/United States
- R56 NS080939/NS/NINDS NIH HHS/United States
- T32 GM007198/GM/NIGMS NIH HHS/United States
- R35 HG011922/HG/NHGRI NIH HHS/United States
- T32 CA067754/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
