

Base editing screens define the genetic landscape of cancer drug resistance mechanisms
- PMID: 39424923
- PMCID: PMC11549056
- DOI: 10.1038/s41588-024-01948-8
Base editing screens define the genetic landscape of cancer drug resistance mechanisms
Abstract
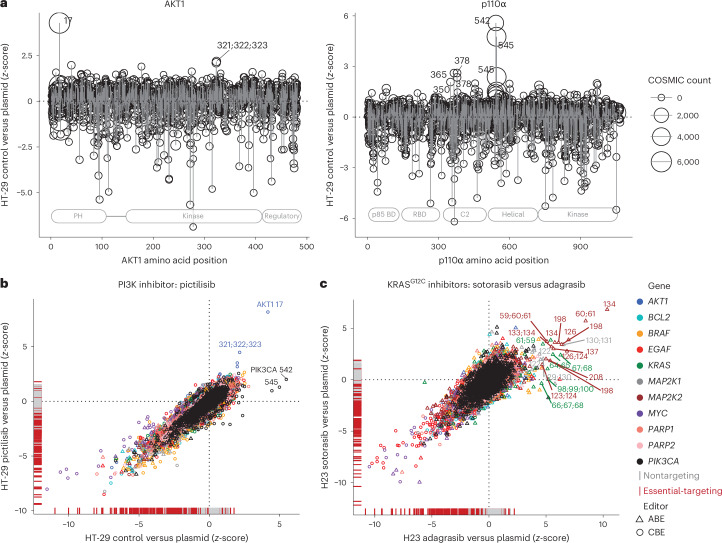
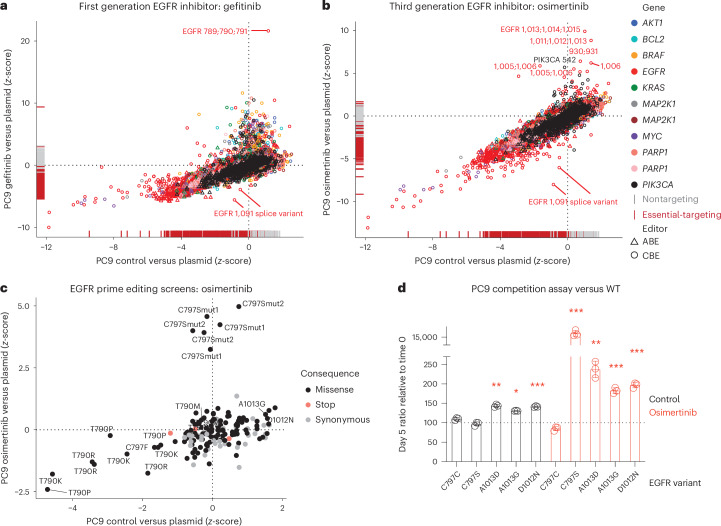
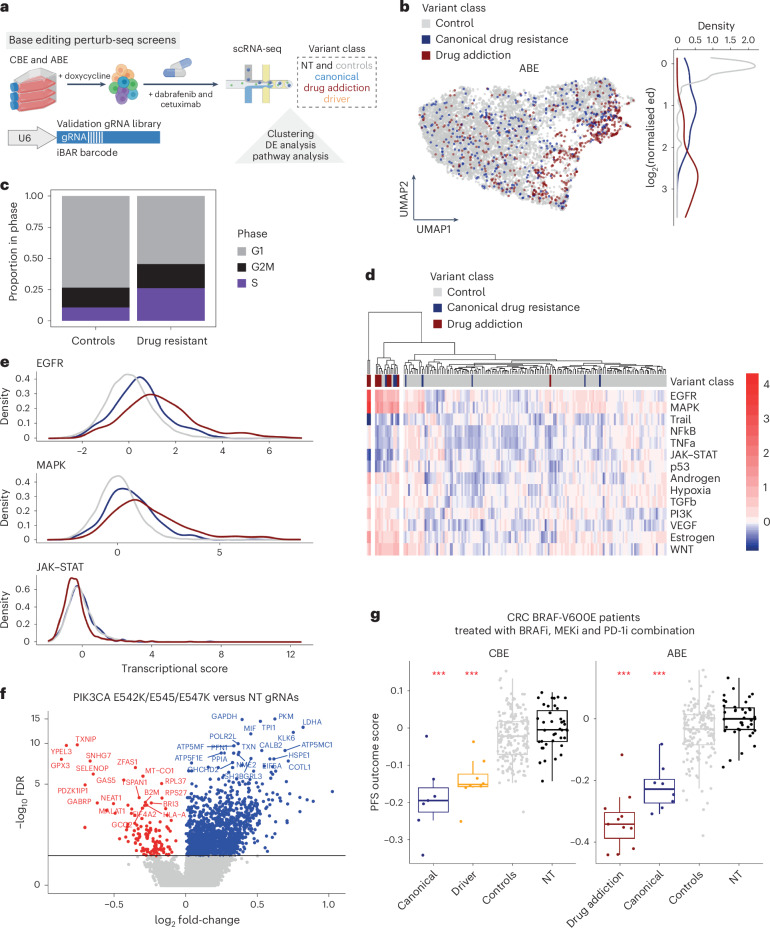
Drug resistance is a principal limitation to the long-term efficacy of cancer therapies. Cancer genome sequencing can retrospectively delineate the genetic basis of drug resistance, but this requires large numbers of post-treatment samples to nominate causal variants. Here we prospectively identify genetic mechanisms of resistance to ten oncology drugs from CRISPR base editing mutagenesis screens in four cancer cell lines using a guide RNA library predicted to install 32,476 variants in 11 cancer genes. We identify four functional classes of protein variants modulating drug sensitivity and use single-cell transcriptomics to reveal how these variants operate through distinct mechanisms, including eliciting a drug-addicted cell state. We identify variants that can be targeted with alternative inhibitors to overcome resistance and functionally validate an epidermal growth factor receptor (EGFR) variant that sensitizes lung cancer cells to EGFR inhibitors. Our variant-to-function map has implications for patient stratification, therapy combinations and drug scheduling in cancer treatment.
© 2024. The Author(s).
Conflict of interest statement
M.J.G. has received research grants from AstraZeneca, GSK and Astex Pharmaceuticals, and is a founder and advisor for Mosaic Therapeutics. J.C.M. has been an employee of Genentech since September 2022. A.B. is a founder and consultant for EnsoCell Therapeutics. M.A.C. and M.J.G. are inventors on patent applications that encompass work described in this paper. J.V.F. and G.I. are employees and shareholders of AstraZeneca. E.E.V. is a founder and advisor for Mosaic Therapeutics. The other authors declare no competing interests.
Figures















References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
