

Interruption of glucagon signaling augments islet non-alpha cell proliferation in SLC7A2- and mTOR-dependent manners
- PMID: 39433176
- PMCID: PMC11570739
- DOI: 10.1016/j.molmet.2024.102050
Interruption of glucagon signaling augments islet non-alpha cell proliferation in SLC7A2- and mTOR-dependent manners
Abstract
Objective: Dysregulated glucagon secretion and inadequate functional beta cell mass are hallmark features of diabetes. While glucagon receptor (GCGR) antagonism ameliorates hyperglycemia and elicits beta cell regeneration in pre-clinical models of diabetes, it also promotes alpha and delta cell hyperplasia. We sought to investigate the mechanism by which loss of glucagon action impacts pancreatic islet non-alpha cells, and the relevance of these observations in a human islet context.
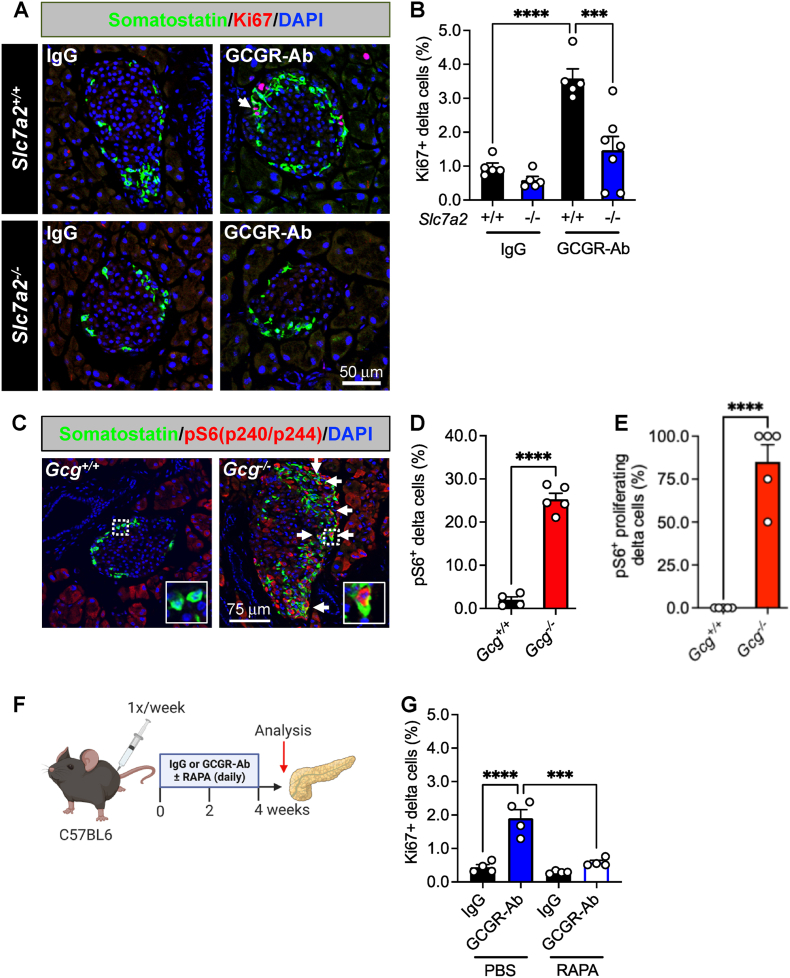
Methods: We used zebrafish, rodents, and transplanted human islets comprising six different models of interrupted glucagon signaling to examine their impact on delta and beta cell proliferation and mass. We also used models with global deficiency of the cationic amino acid transporter, SLC7A2, and mTORC1 inhibition via rapamycin, to determine whether amino acid-dependent nutrient sensing was required for islet non-alpha cell growth.
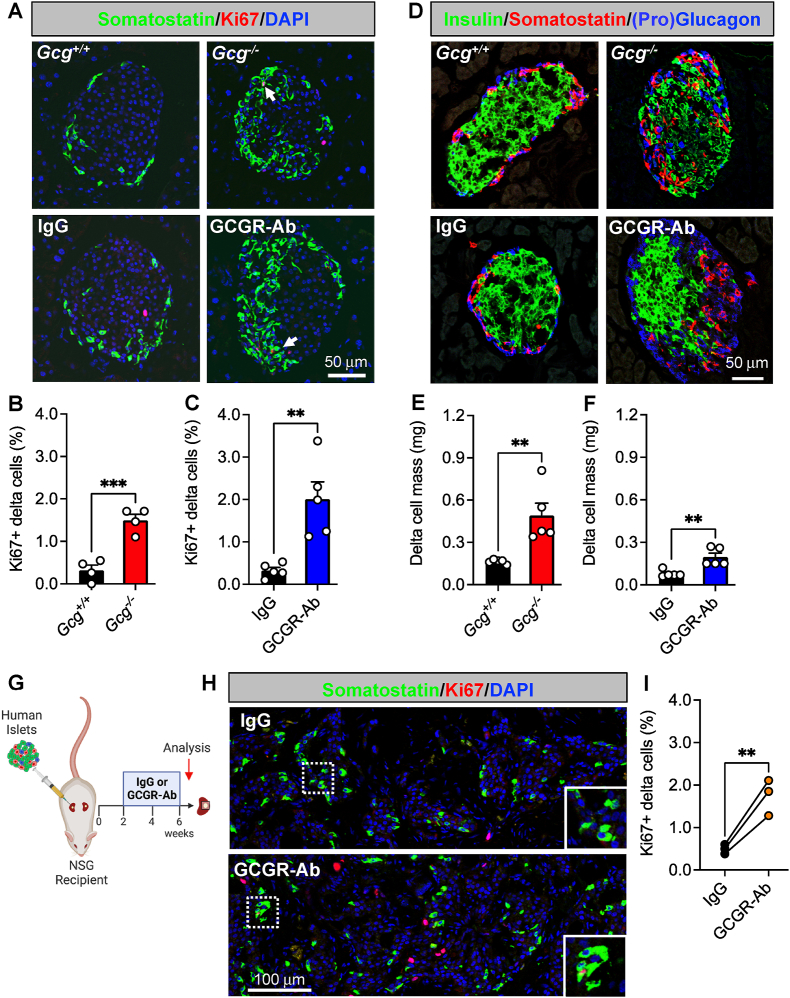
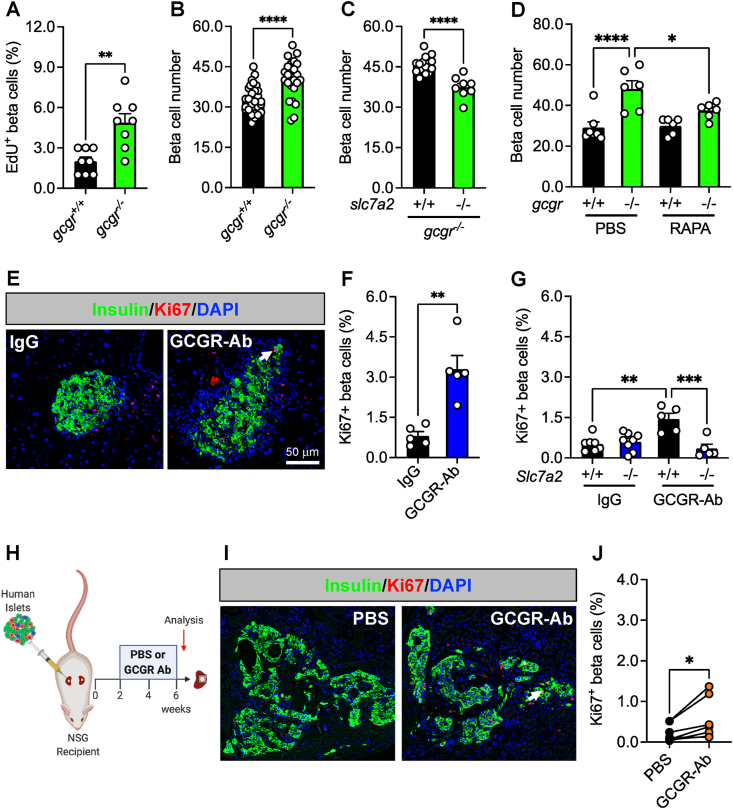
Results: Inhibition of glucagon signaling stimulated delta cell proliferation in mouse and transplanted human islets, and in mouse islets. This was rapamycin-sensitive and required SLC7A2. Likewise, gcgr deficiency augmented beta cell proliferation via SLC7A2- and mTORC1-dependent mechanisms in zebrafish and promoted cell cycle engagement in rodent beta cells but was insufficient to drive a significant increase in beta cell mass in mice.
Conclusions: Our findings demonstrate that interruption of glucagon signaling augments islet non-alpha cell proliferation in zebrafish, rodents, and transplanted human islets in a manner requiring SLC7A2 and mTORC1 activation. An increase in delta cell mass may be leveraged for future beta cell regeneration therapies relying upon delta cell reprogramming.
Keywords: Beta cell; Delta cell; Glucagon receptor; Pancreatic islet; Proliferation; mTOR.
Copyright © 2024 The Authors. Published by Elsevier GmbH.. All rights reserved.
Conflict of interest statement
Declaration of competing interest Hai Yan is an employee of REMD Biotherapeutics Inc. which licensed one of the glucagon receptor antibodies used for some of the studies in this manuscript. All other authors have no interests to declare.
Figures
Update of
-
Interruption of glucagon signaling augments islet non-alpha cell proliferation in SLC7A2- and mTOR-dependent manners.bioRxiv [Preprint]. 2024 Aug 7:2024.08.06.606926. doi: 10.1101/2024.08.06.606926. bioRxiv. 2024. Update in: Mol Metab. 2024 Dec;90:102050. doi: 10.1016/j.molmet.2024.102050. PMID: 39149351 Free PMC article. Updated. Preprint.
References
-
- Muller W.A., Faloona G.R., Aguilar-Parada E., Unger R.H. Abnormal alpha-cell function in diabetes. Response to carbohydrate and protein ingestion. N Engl J Med. 1970;283(3):109–115. - PubMed
-
- Unger R., Orci L. The essential role of glucagon in the pathogenesis of diabetes mellitus. Lancet. 1975;305(7897):14–16. - PubMed
-
- Kahn S.E. The importance of β-cell failure in the development and progression of type 2 diabetes. J Clin Endocrinol Metabol. 2001;86(9):4047–4058. - PubMed
-
- Dunning B.E., Gerich J.E. The role of alpha-cell dysregulation in fasting and postprandial hyperglycemia in type 2 diabetes and therapeutic implications. Endocr Rev. 2007;28(3):253–283. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
