

Harnessing the power of AI in precision medicine: NGS-based therapeutic insights for colorectal cancer cohort
- PMID: 39435285
- PMCID: PMC11491396
- DOI: 10.3389/fonc.2024.1407465
Harnessing the power of AI in precision medicine: NGS-based therapeutic insights for colorectal cancer cohort
Abstract
Purpose: Developing innovative precision and personalized cancer therapeutics is essential to enhance cancer survivability, particularly for prevalent cancer types such as colorectal cancer. This study aims to demonstrate various approaches for discovering new targets for precision therapies using artificial intelligence (AI) on a Polish cohort of colorectal cancer patients.
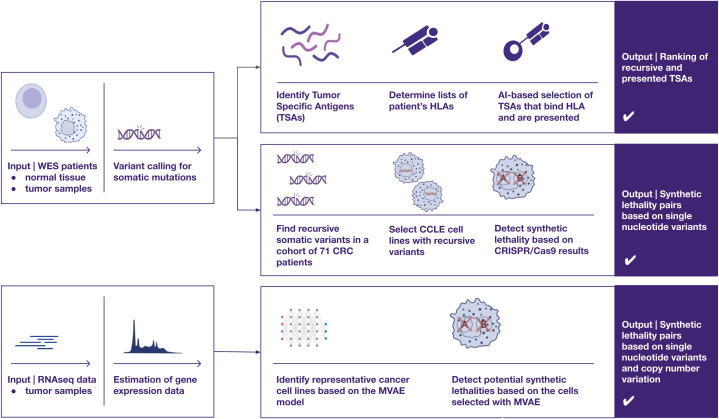
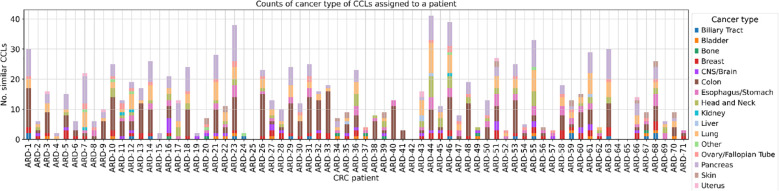
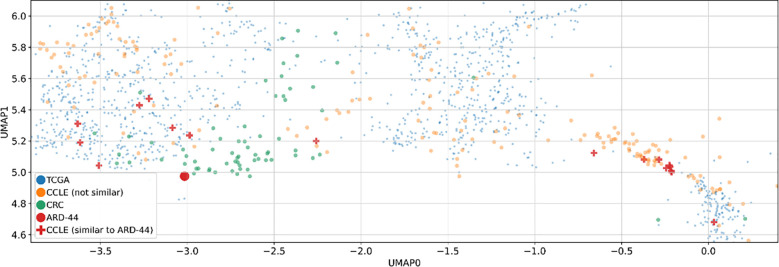
Methods: We analyzed 71 patients with histopathologically confirmed advanced resectional colorectal adenocarcinoma. Whole exome sequencing was performed on tumor and peripheral blood samples, while RNA sequencing (RNAseq) was conducted on tumor samples. We employed three approaches to identify potential targets for personalized and precision therapies. First, using our in-house neoantigen calling pipeline, ARDentify, combined with an AI-based model trained on immunopeptidomics mass spectrometry data (ARDisplay), we identified neoepitopes in the cohort. Second, based on recurrent mutations found in our patient cohort, we selected corresponding cancer cell lines and utilized knock-out gene dependency scores to identify synthetic lethality genes. Third, an AI-based model trained on cancer cell line data was employed to identify cell lines with genomic profiles similar to selected patients. Copy number variants and recurrent single nucleotide variants in these cell lines, along with gene dependency data, were used to find personalized synthetic lethality pairs.
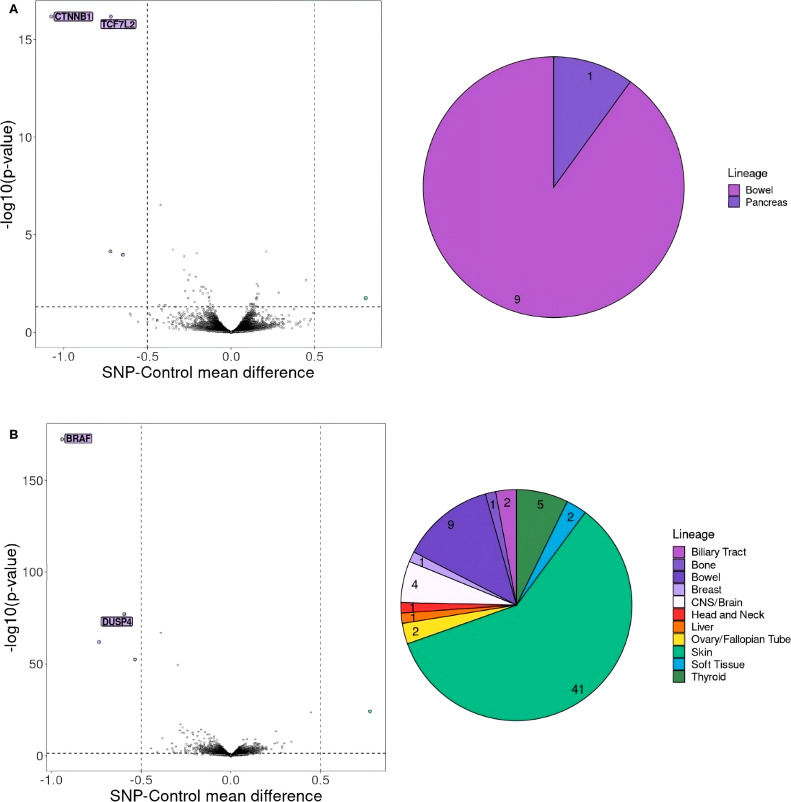
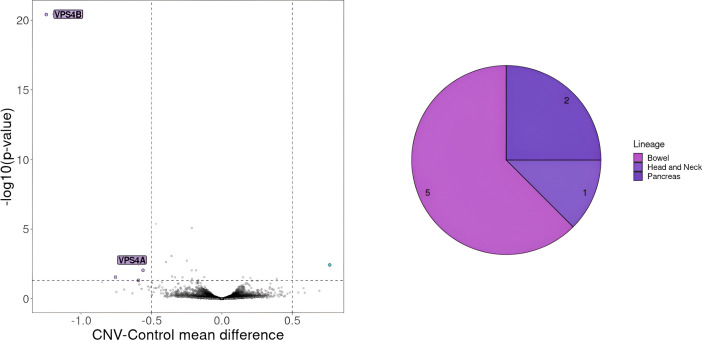
Results: We identified approximately 8,700 unique neoepitopes, but none were shared by more than two patients, indicating limited potential for shared neoantigenic targets across our cohort. Additionally, we identified three synthetic lethality pairs: the well-known APC-CTNNB1 and BRAF-DUSP4 pairs, along with the recently described APC-TCF7L2 pair, which could be significant for patients with APC and BRAF variants. Furthermore, by leveraging the identification of similar cancer cell lines, we uncovered a potential gene pair, VPS4A and VPS4B, with therapeutic implications.
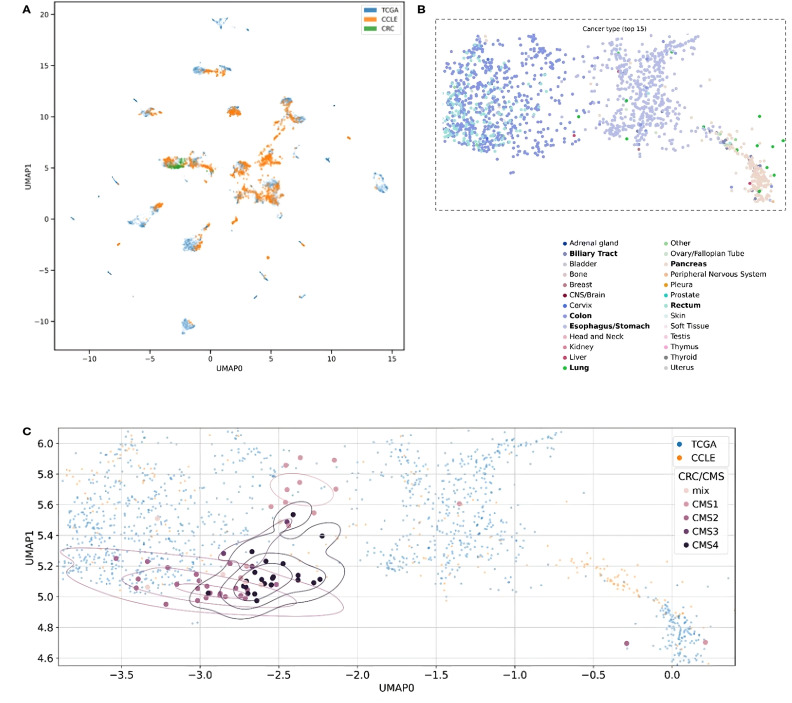
Conclusion: Our study highlights three distinct approaches for identifying potential therapeutic targets in cancer patients. Each approach yielded valuable insights into our cohort, underscoring the relevance and utility of these methodologies in the development of precision and personalized cancer therapies. Importantly, we developed a novel AI model that aligns tumors with representative cell lines using RNAseq and methylation data. This model enables us to identify cell lines closely resembling patient tumors, facilitating accurate selection of models needed for in vitro validation.
Keywords: AI; CRC; neoantigens; precision medicine; synthetic lethality.
Copyright © 2024 Murcia Pienkowski, Skoczylas, Zaremba, Kłęk, Balawejder, Biernat, Czarnocka, Gniewek, Grochowalski, Kamuda, Król-Józaga, Marczyńska-Grzelak, Mazzocco, Szatanek, Widawski, Welanyk, Orzeszko, Szura, Torbicz, Borys, Wohadlo, Wysocki, Karczewski, Markowska, Kucharczyk, Piatek, Jasiński, Warchoł, Kaczmarczyk, Blum and Sanecka-Duin.
Conflict of interest statement
Authors VMP, PS, AZ, MBa, PB, WC, OG, ŁG, MKam, BK-J, JM-G, GM, RS, JWi, MP, MJ, MWa, JK and AB were employed by Ardigen SA. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous