

Enriched Environment Reduces Seizure Susceptibility via Entorhinal Cortex Circuit Augmented Adult Neurogenesis
- PMID: 39435757
- PMCID: PMC11633471
- DOI: 10.1002/advs.202410927
Enriched Environment Reduces Seizure Susceptibility via Entorhinal Cortex Circuit Augmented Adult Neurogenesis
Abstract
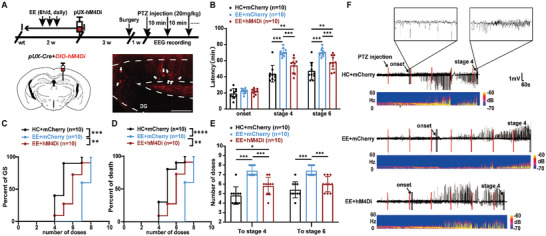
Enriched environment (EE), characterized by multi-sensory stimulation, represents a non-invasive alternative for alleviating epileptic seizures. However, the mechanism by which EE exerts its therapeutic impact remains incompletely understood. Here, it is elucidated that EE mitigates seizure susceptibility through the augmentation of adult neurogenesis within the entorhinal cortex (EC) circuit. A substantial upregulation of adult hippocampal neurogenesis concomitant with a notable reduction in seizure susceptibility has been found following exposure to EE. EE-enhanced adult-born dentate granule cells (abDGCs) are functionally activated during seizure events. Importantly, the selective activation of abDGCs mimics the anti-seizure effects observed with EE, while their inhibition negates these effects. Further, whole-brain c-Fos mapping demonstrates increased activity in DG-projecting EC CaMKIIα+ neurons in response to EE. Crucially, EC CaMKIIα+ neurons exert bidirectional modulation over the proliferation and maturation of abDGCs that can activate local GABAergic interneurons; thus, they are essential components for the anti-seizure effects mediated by EE. Collectively, this study provides compelling evidence regarding the circuit mechanisms underlying the effects of EE treatment on epileptic seizures, shedding light on the involvement of the EC-DG circuit in augmenting the functionality of abDGCs. This may help for the translational application of EE for epilepsy management.
Keywords: EC‐DG pathway; enriched environment; epilepsy; neurogenesis.
© 2024 The Author(s). Advanced Science published by Wiley‐VCH GmbH.
Conflict of interest statement
The authors declare no conflict of interest.
Figures






References
-
- a) Jetté N., Sander J. W., Keezer M. R., Lancet Neurol. 2016, 15, 982; - PubMed
- b) Chen J., Li Z., Chen L., Acta Epileptologica. 2022, 4, 35.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases