

Evaluation of Alpha-Synuclein and Tau Antiaggregation Activity of Urea and Thiourea-Based Small Molecules for Neurodegenerative Disease Therapeutics
- PMID: 39436010
- PMCID: PMC11587513
- DOI: 10.1021/acschemneuro.4c00282
Evaluation of Alpha-Synuclein and Tau Antiaggregation Activity of Urea and Thiourea-Based Small Molecules for Neurodegenerative Disease Therapeutics
Abstract
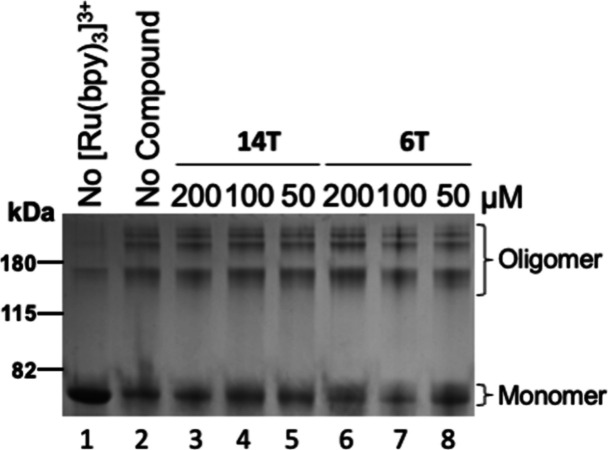
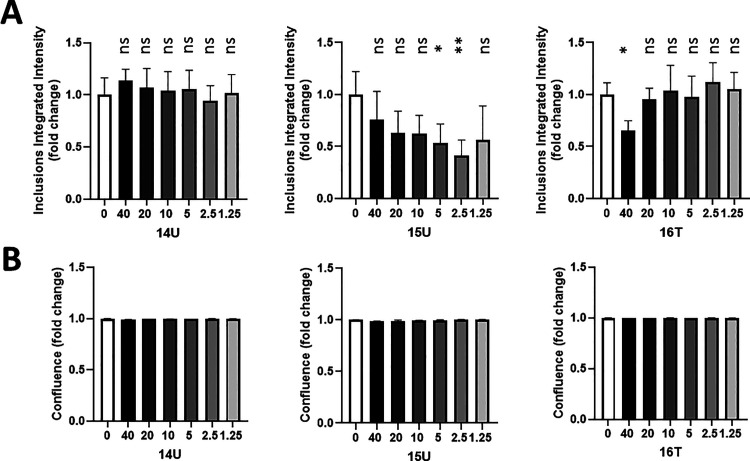
Alzheimer's disease (AD) and Parkinson's disease (PD) are multifactorial, chronic diseases involving neurodegeneration. According to recent studies, it is hypothesized that the intraneuronal and postsynaptic accumulation of misfolded proteins such as α-synuclein (α-syn) and tau, responsible for Lewy bodies (LB) and tangles, respectively, disrupts neuron functions. Considering the co-occurrence of α-syn and tau inclusions in the brains of patients afflicted with subtypes of dementia and LB disorders, the discovery and development of small molecules for the inhibition of α-syn and tau aggregation can be a potentially effective strategy to delay neurodegeneration. Urea is a chaotropic agent that alters protein solubilization and hydrophobic interactions and inhibits protein aggregation and precipitation. The presence of three hetero atoms (O/S and N) in proximity can coordinate with neutral, mono, or dianionic groups to form stable complexes in the biological system. Therefore, in this study, we evaluated urea and thiourea linkers with various substitutions on either side of the carbamide or thiocarbamide functionality to compare the aggregation inhibition of α-syn and tau. A thioflavin-T (ThT) fluorescence assay was used to evaluate the level of fibril formation and monitor the anti-aggregation effect of the different compounds. We opted for transmission electron microscopy (TEM) as a direct means to confirm the anti-fibrillar effect. The oligomer formation was monitored via the photoinduced cross-linking of unmodified proteins (PICUP). The anti-inclusion and anti-seeding activities of the best compounds were evaluated using M17D intracellular inclusion and biosensor cell-based assays, respectively. Disaggregation experiments were performed with amyloid plaques extracted from AD brains. The analogues with indole, benzothiazole, or N,N-dimethylphenyl on one side with halo-substituted aromatic moieties had shown less than 15% cutoff fluorescence obtained with the ThT assay. Our lead molecules 6T and 14T reduced α-syn oligomerization dose-dependently based on the PICUP assays but failed at inhibiting tau oligomer formation. The anti-inclusion effect of our lead compounds was confirmed using the M17D neuroblastoma cell model. Compounds 6T and 14T exhibited an anti-seeding effect on tau using biosensor cells. In contrast to the control, disaggregation experiments showed fewer Aβ plaques with our lead molecules (compounds 6T and 14T). Pharmacokinetics (PK) mice studies demonstrated that these two thiourea-based small molecules have the potential to cross the blood-brain barrier in rodents. Urea and thiourea linkers could be further improved for their PK parameters and studied for the anti-inclusion, anti-seeding, and disaggregation effects using transgenic mice models of neurodegenerative diseases.
Keywords: Alzheimer’s disease; Parkinson’s disease; anti-aggregation; neurodegeneration; thiourea; urea.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
















References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
