

FoxO1/Rictor axis induces a nongenetic adaptation to ibrutinib via Akt activation in chronic lymphocytic leukemia
- PMID: 39436708
- PMCID: PMC11601945
- DOI: 10.1172/JCI173770
FoxO1/Rictor axis induces a nongenetic adaptation to ibrutinib via Akt activation in chronic lymphocytic leukemia
Abstract
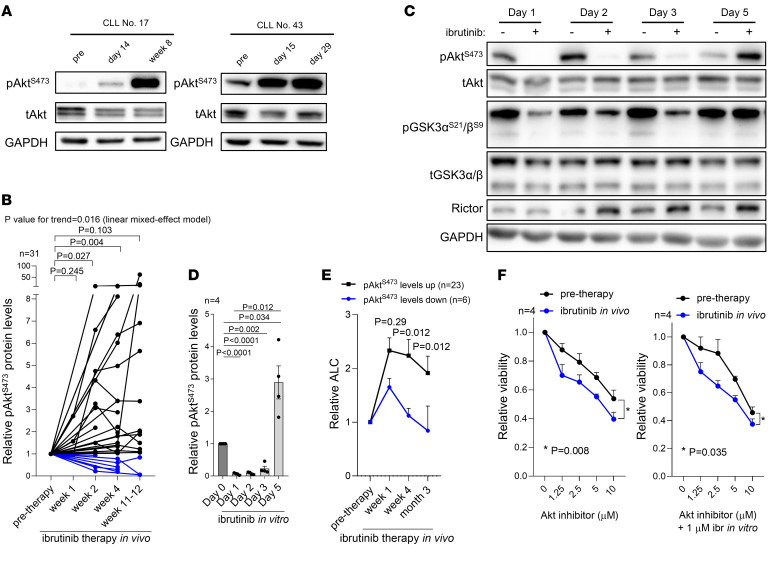
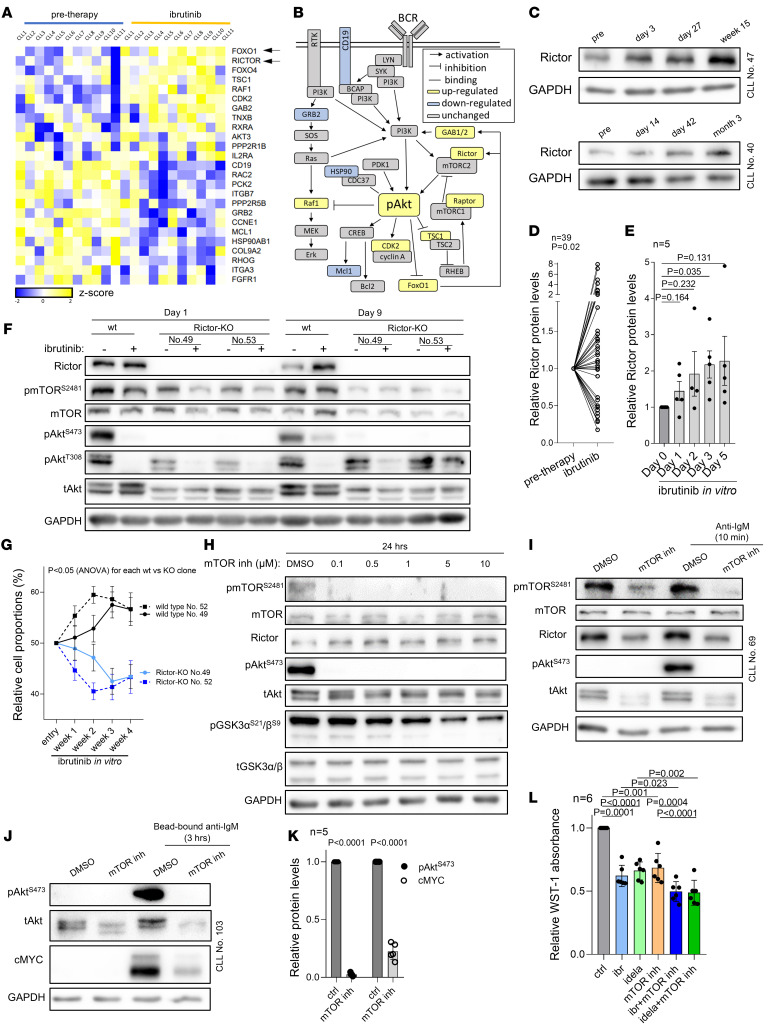
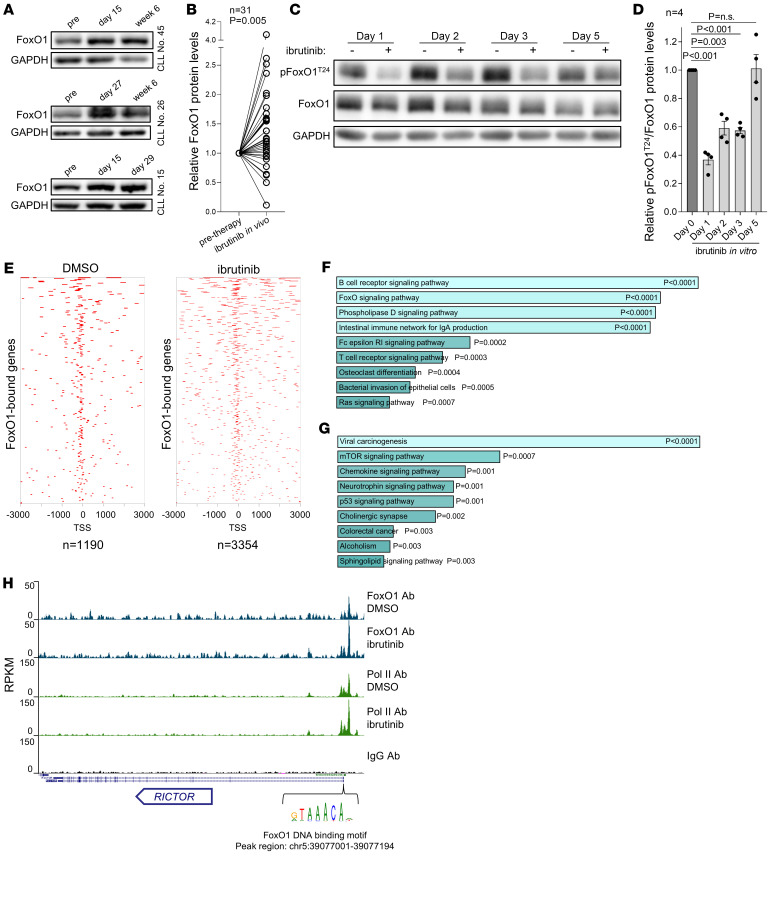
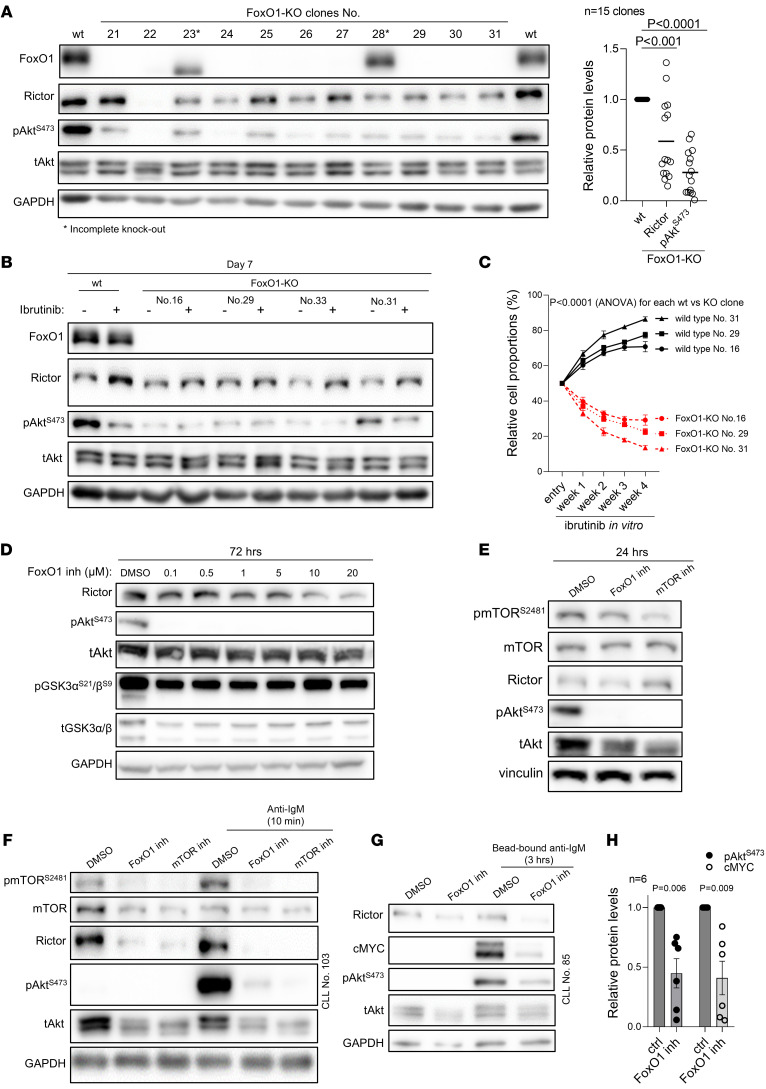
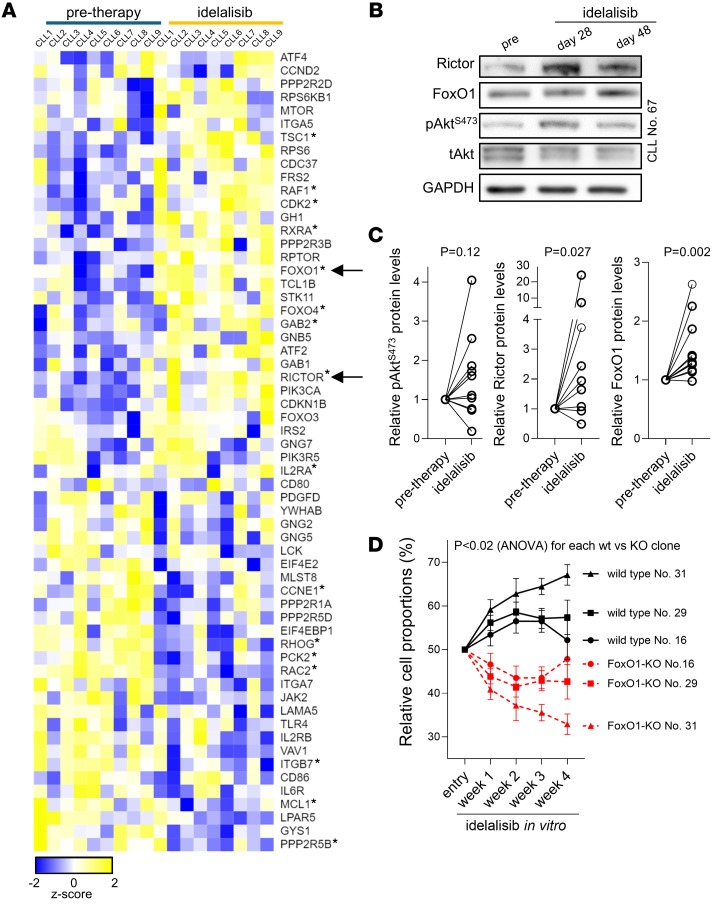
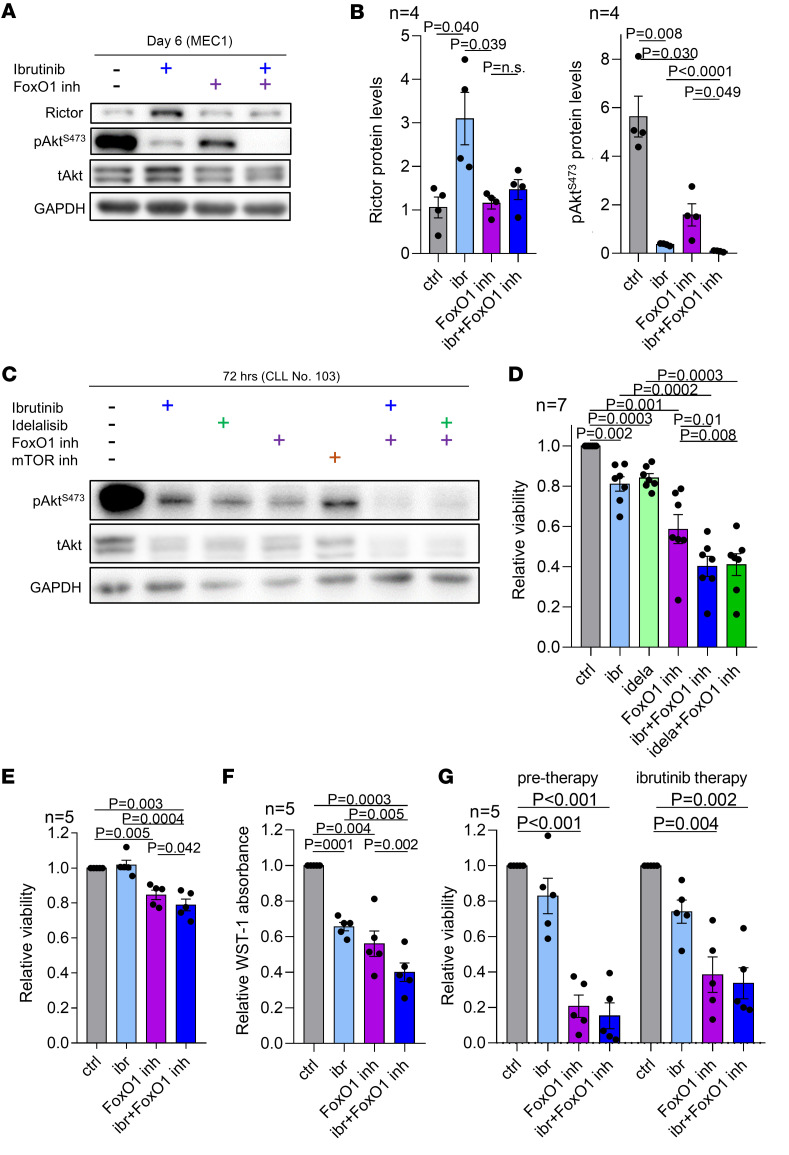
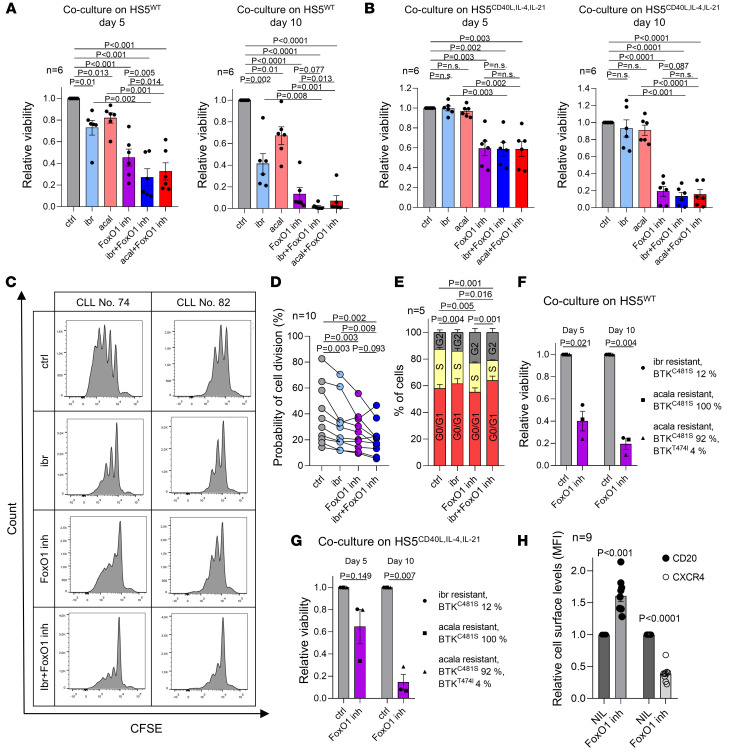
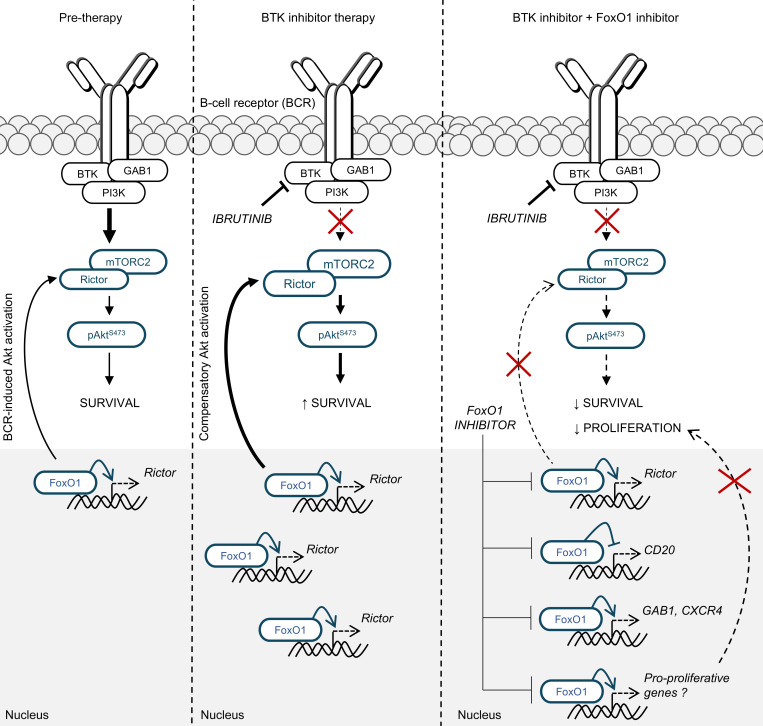
Bruton tyrosine kinase (BTK) inhibitor therapy induces peripheral blood lymphocytosis in chronic lymphocytic leukemia (CLL), which lasts for several months. It remains unclear whether nongenetic adaptation mechanisms exist, allowing CLL cells' survival during BTK inhibitor-induced lymphocytosis and/or playing a role in therapy resistance. We show that in approximately 70% of CLL cases, ibrutinib treatment in vivo increases Akt activity above pretherapy levels within several weeks, leading to compensatory CLL cell survival and a more prominent lymphocytosis on therapy. Ibrutinib-induced Akt phosphorylation (pAktS473) is caused by the upregulation of Forkhead box protein O1 (FoxO1) transcription factor, which induces expression of Rictor, an assembly protein for the mTORC2 protein complex that directly phosphorylates Akt at serine 473 (S473). Knockout or inhibition of FoxO1 or Rictor led to a dramatic decrease in Akt phosphorylation and growth disadvantage for malignant B cells in the presence of ibrutinib (or PI3K inhibitor idelalisib) in vitro and in vivo. The FoxO1/Rictor/pAktS473 axis represents an early nongenetic adaptation to B cell receptor (BCR) inhibitor therapy not requiring PI3Kδ or BTK kinase activity. We further demonstrate that FoxO1 can be targeted therapeutically and its inhibition induces CLL cells' apoptosis alone or in combination with BTK inhibitors (ibrutinib, acalabrutinib, pirtobrutinib) and blocks their proliferation triggered by T cell factors (CD40L, IL-4, and IL-21).
Keywords: Drug therapy; Hematology; Leukemias; Oncology; Signal transduction.
Conflict of interest statement
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
