

Neurodevelopmental Disorder Caused by Deletion of CHASERR, a lncRNA Gene
- PMID: 39442041
- PMCID: PMC11826417
- DOI: 10.1056/NEJMoa2400718
Neurodevelopmental Disorder Caused by Deletion of CHASERR, a lncRNA Gene
Abstract
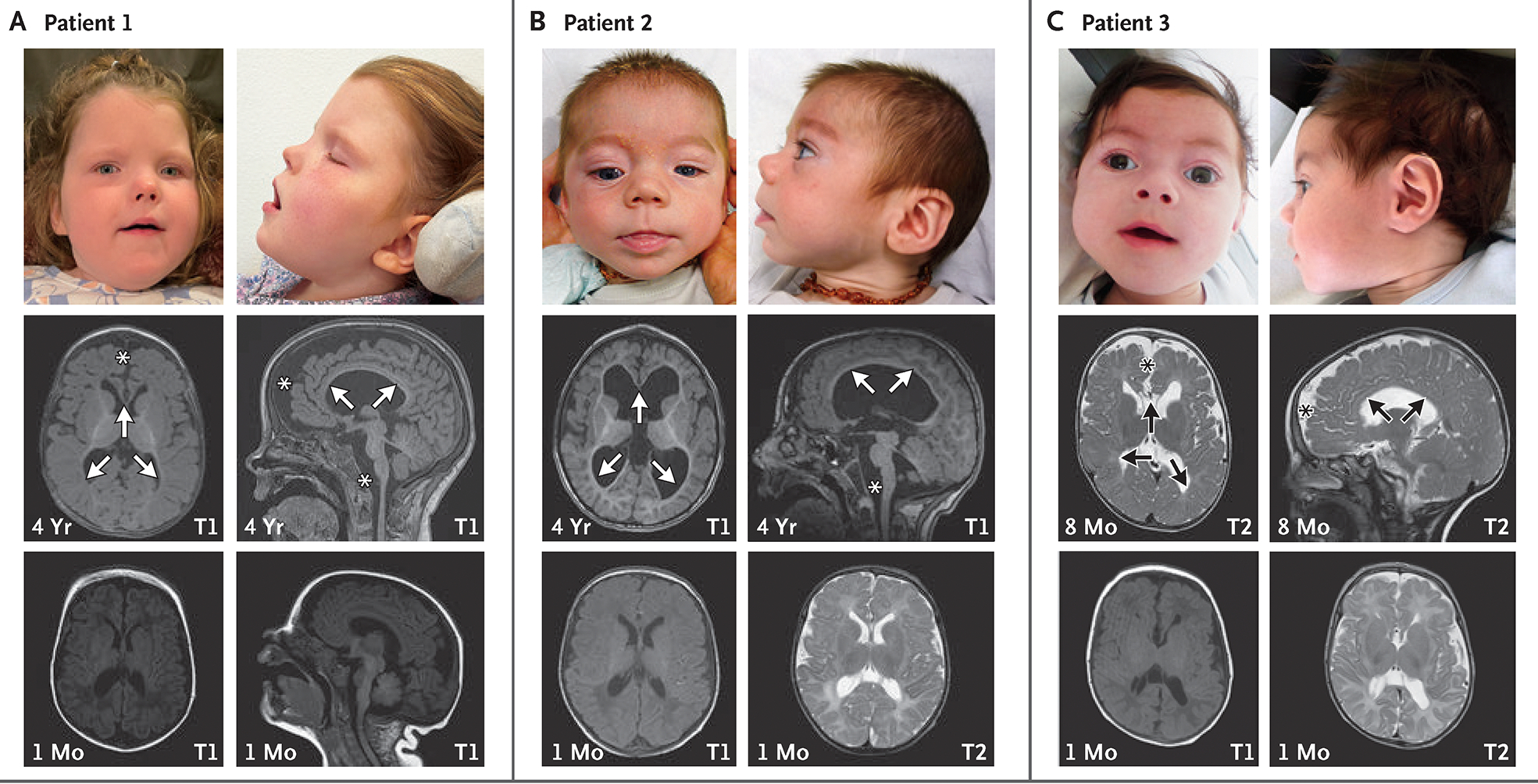
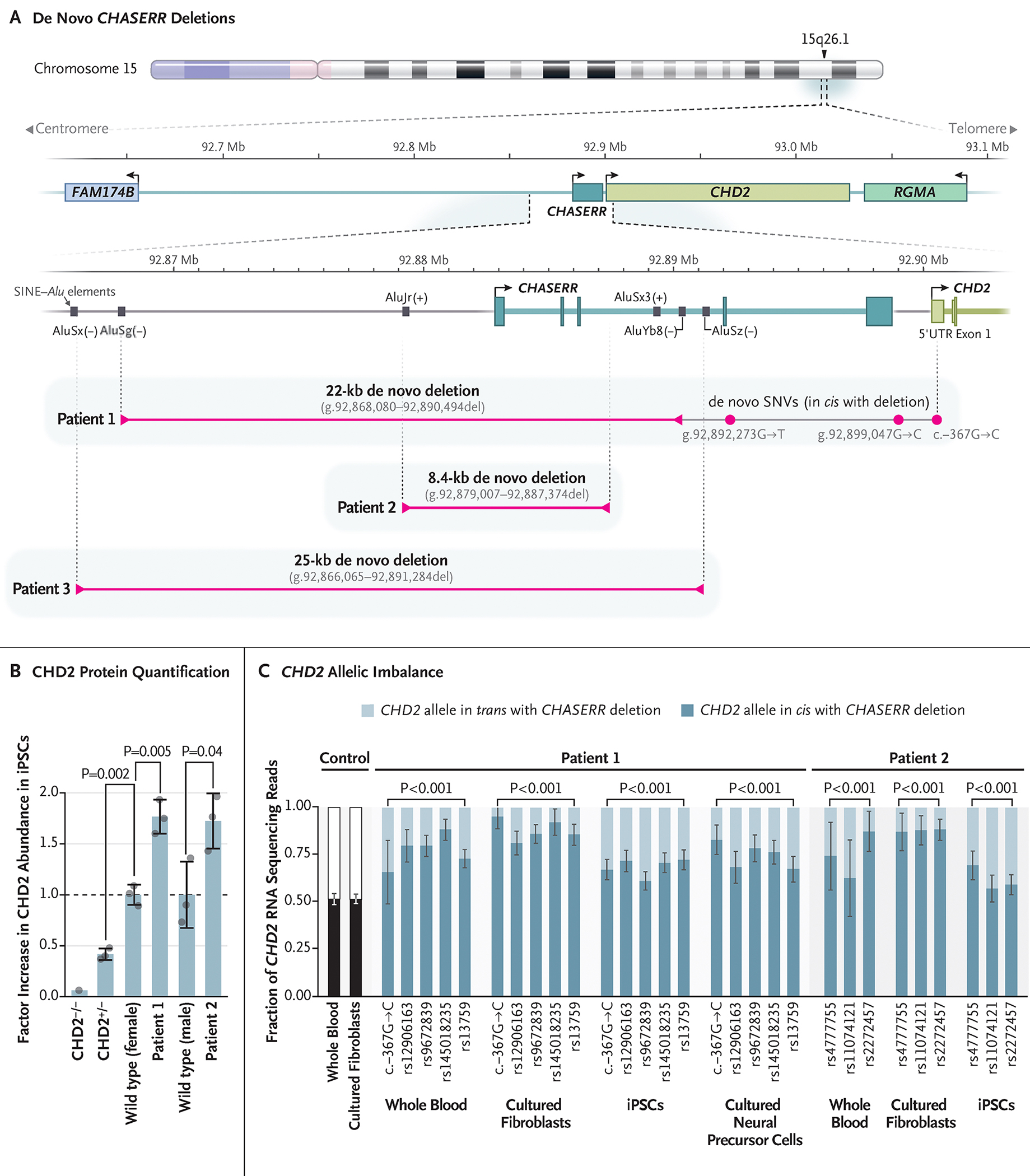
CHASERR encodes a human long noncoding RNA (lncRNA) adjacent to CHD2, a coding gene in which de novo loss-of-function variants cause developmental and epileptic encephalopathy. Here, we report our findings in three unrelated children with a syndromic, early-onset neurodevelopmental disorder, each of whom had a de novo deletion in the CHASERR locus. The children had severe encephalopathy, shared facial dysmorphisms, cortical atrophy, and cerebral hypomyelination - a phenotype that is distinct from the phenotypes of patients with CHD2 haploinsufficiency. We found that the CHASERR deletion results in increased CHD2 protein abundance in patient-derived cell lines and increased expression of the CHD2 transcript in cis. These findings indicate that CHD2 has bidirectional dosage sensitivity in human disease, and we recommend that other lncRNA-encoding genes be evaluated, particularly those upstream of genes associated with mendelian disorders. (Funded by the National Human Genome Research Institute and others.).
Copyright © 2024 Massachusetts Medical Society.
Figures
References
-
- Shen T, Ji F, Yuan Z, Jiao J. CHD2 is required for embryonic neurogenesis in the developing cerebral cortex. Stem Cells 2015;33:1794–806. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- UM1 HG008900/HG/NHGRI NIH HHS/United States
- U01HG007943/Common Fund
- 2019-199278 (funder DOI 10.13039/100014989)/Chan Zuckerberg Initiative
- U01 HG011755/HG/NHGRI NIH HHS/United States
- UM1HG008900/HG/NHGRI NIH HHS/United States
- U01HG011755/HG/NHGRI NIH HHS/United States
- K23AR083505-01/AR/NIAMS NIH HHS/United States
- R01 HG009141/HG/NHGRI NIH HHS/United States
- U01HG007530/Common Fund
- U01HG007709/Common Fund
- K23 AR083505/AR/NIAMS NIH HHS/United States
- T32 GM142604/GM/NIGMS NIH HHS/United States
- F32 HD101280/HD/NICHD NIH HHS/United States
- R01HG009141/HG/NHGRI NIH HHS/United States
- R00 NS089858/NS/NINDS NIH HHS/United States
- T32 HG10464/HG/NHGRI NIH HHS/United States
- K99/R00NS089858/NS/NINDS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Molecular Biology Databases