

Molecular Phenotypes Segregate Missense Mutations in SLC13A5 Epilepsy
- PMID: 39442909
- PMCID: PMC11929159
- DOI: 10.1016/j.jmb.2024.168820
Molecular Phenotypes Segregate Missense Mutations in SLC13A5 Epilepsy
Abstract
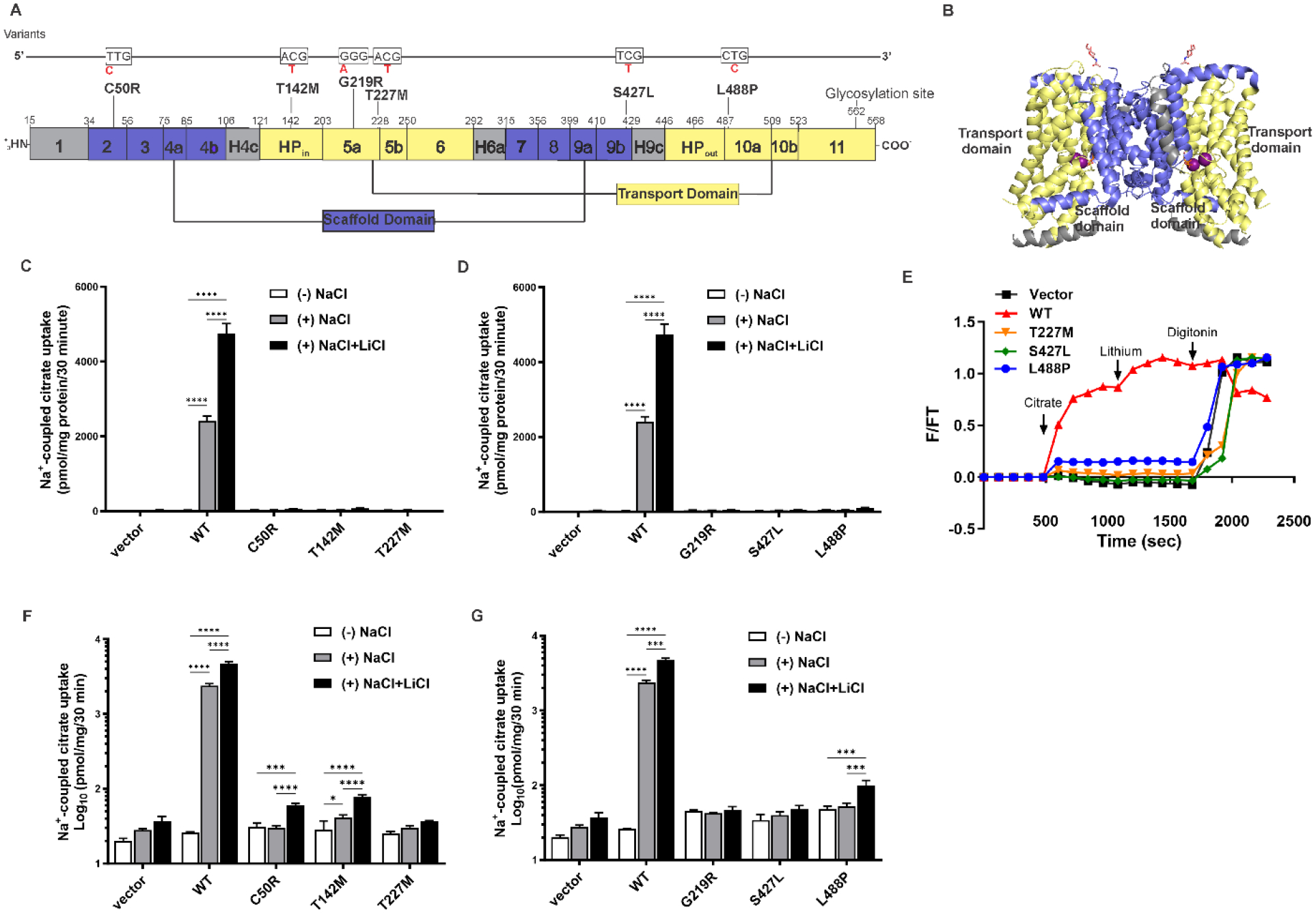
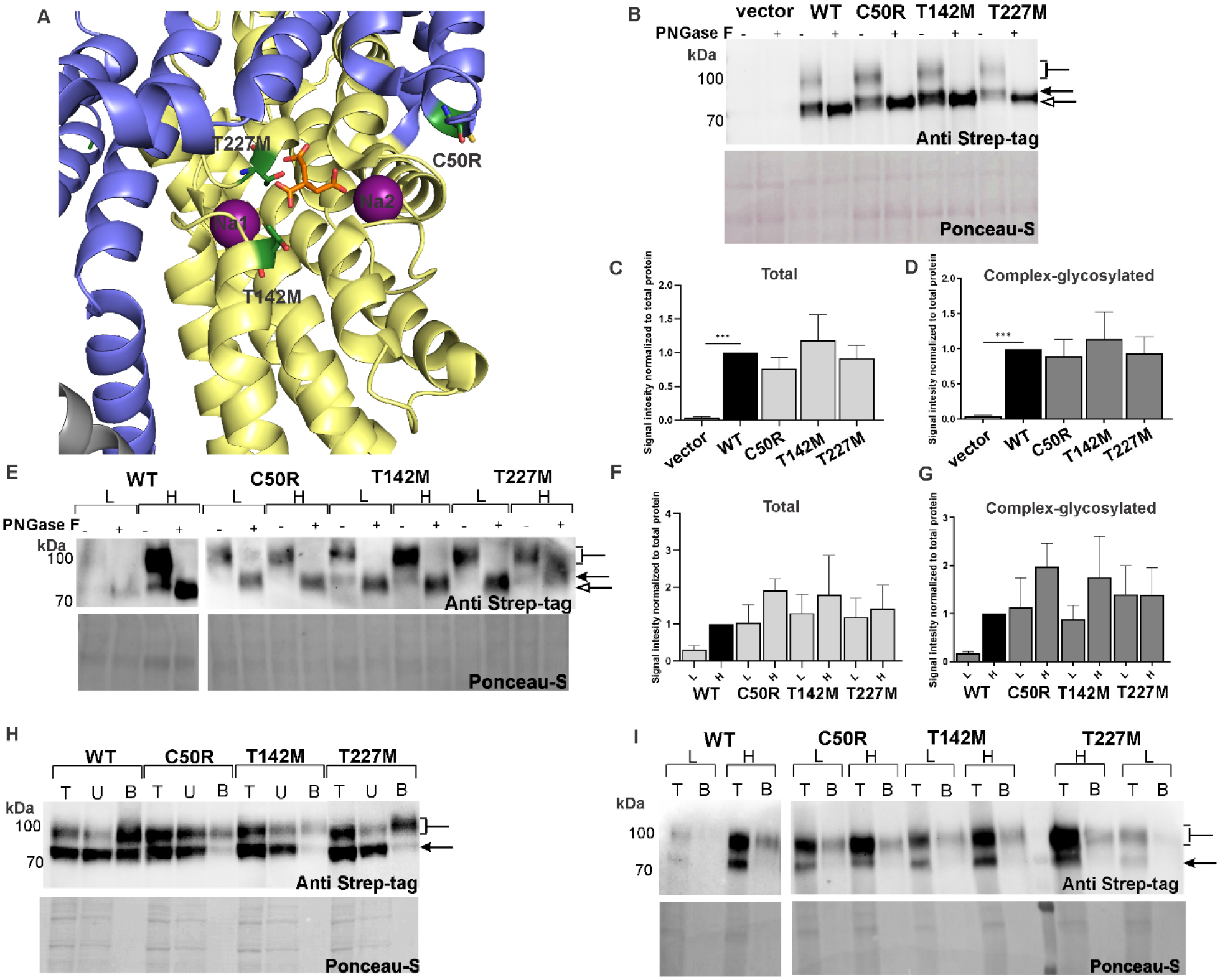
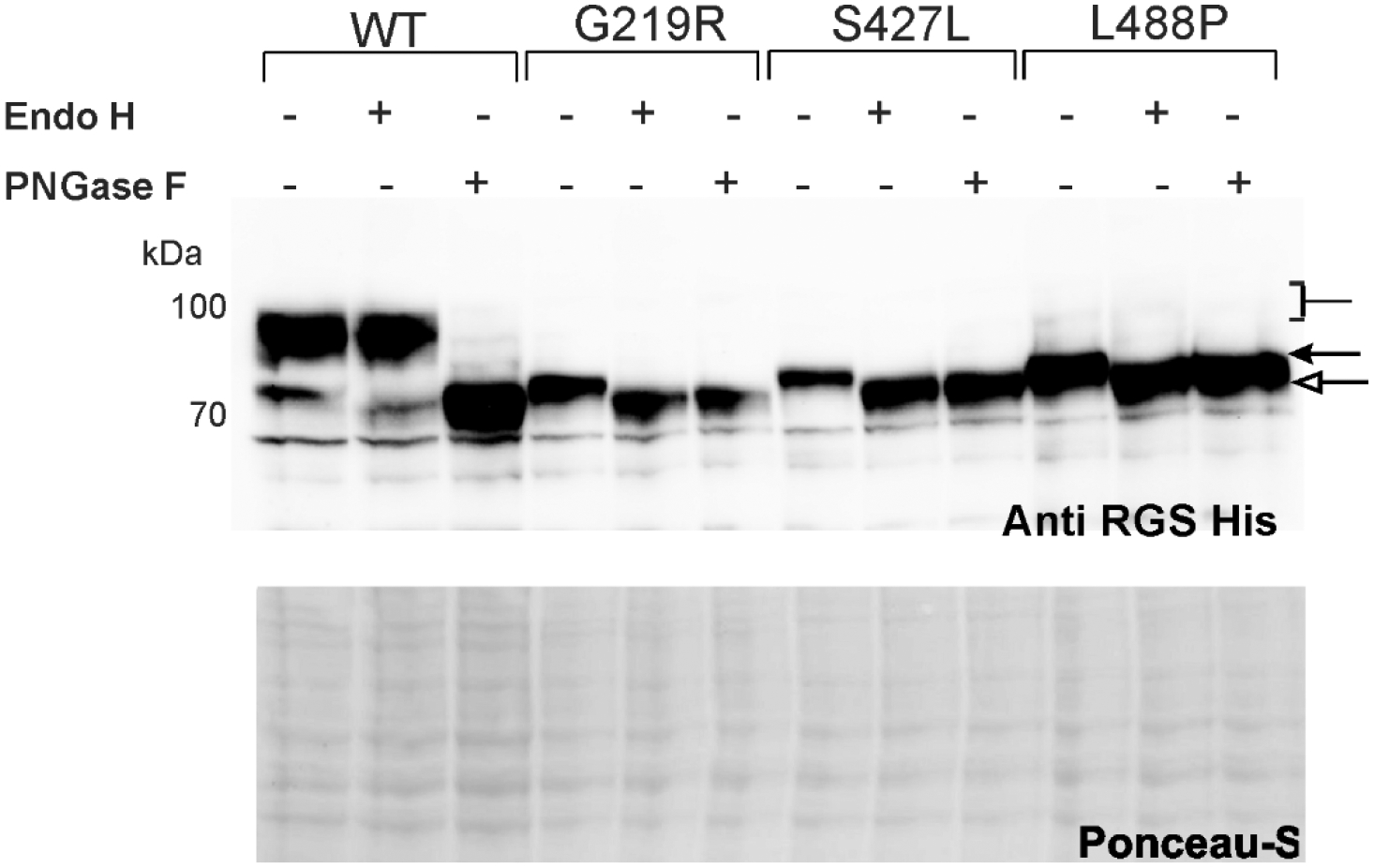
The sodium-coupled citrate transporter (NaCT, SLC13A5) mediates citrate uptake across the plasma membrane via an inward Na+ gradient. Mutations in SLC13A5 cause early infantile epileptic encephalopathy type-25 (EIEE25, SLC13A5 Epilepsy) due to impaired citrate uptake in neurons and astrocytes. Despite clinical identification of disease-causing mutations, underlying mechanisms and cures remain elusive. Here we mechanistically classify six frequent SLC13A5 mutations by phenotyping their protein cell surface expression and citrate transport functions. Mutants C50R, T142M, and T227M exhibit impaired citrate transport despite normal expression at the cell surface. In contrast, mutations G219R, S427L, and L488P show low total protein expression levels, absence of mature, glycosylated proteins at the cell surface, retention of the proteins in the endoplasmic reticulum, and diminished transport activity. This mechanistic classification divides SLC13A5 mutants into two groups, Class I (C50R, T142M, and T227M) and Class II (G219R, S427L, and L488P). Importantly, mutants' mRNA levels resemble wildtype, suggesting post-translational defects. Class II mutations display immature core-glycosylation and shortened half-lives, indicating protein folding defects. Together, these experiments provide a comprehensive understanding of the disease-causing mutation's defects in SLC13A5 Epilepsy at the biochemical and molecular level and shed light into the trafficking pathway(s) of NaCT. The two classes of mutations will require fundamentally different approaches for treatment to either restore transport function of the mutant protein that is capable of reaching the cell surface (Class I), or therapies that enable the correction of protein folding defects to enable escape to the cell surface where it may restore transport function (Class II).
Keywords: NaCT; biogenesis; folding defect; proteasomal degradation; transport defect.
Copyright © 2024 Elsevier Ltd. All rights reserved.
Conflict of interest statement
Declaration of competing interest The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures






Update of
-
Molecular phenotypes segregate missense mutations in SLC13A5 Epilepsy.bioRxiv [Preprint]. 2024 May 23:2024.05.23.594637. doi: 10.1101/2024.05.23.594637. bioRxiv. 2024. Update in: J Mol Biol. 2024 Nov 15;436(22):168820. doi: 10.1016/j.jmb.2024.168820. PMID: 38826402 Free PMC article. Updated. Preprint.
References
-
- Tong W-H, Rouault TA. Metabolic regulation of citrate and iron by aconitases: role of iron–sulfur cluster biogenesis. Biometals. 2007;20:549–64. - PubMed
-
- Fraenkl SA, Muser J, Groell R, Reinhard G, Orgul S, Flammer J, et al. Plasma citrate levels as a potential biomarker for glaucoma. Journal of ocular pharmacology and therapeutics. 2011;27:577–80. - PubMed
-
- Hamm LL. Renal handling of citrate. Kidney international. 1990;38:728–35. - PubMed
-
- Yodoya E, Wada M, Shimada A, Katsukawa H, Okada N, Yamamoto A, et al. Functional and molecular identification of sodium-coupled dicarboxylate transporters in rat primary cultured cerebrocortical astrocytes and neurons. Journal of neurochemistry. 2006;97:162–73. - PubMed
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
