

Accurate bacterial outbreak tracing with Oxford Nanopore sequencing and reduction of methylation-induced errors
- PMID: 39443152
- PMCID: PMC11610573
- DOI: 10.1101/gr.278848.123
Accurate bacterial outbreak tracing with Oxford Nanopore sequencing and reduction of methylation-induced errors
Abstract
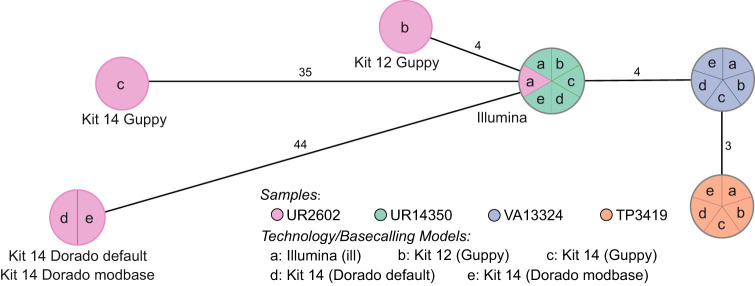
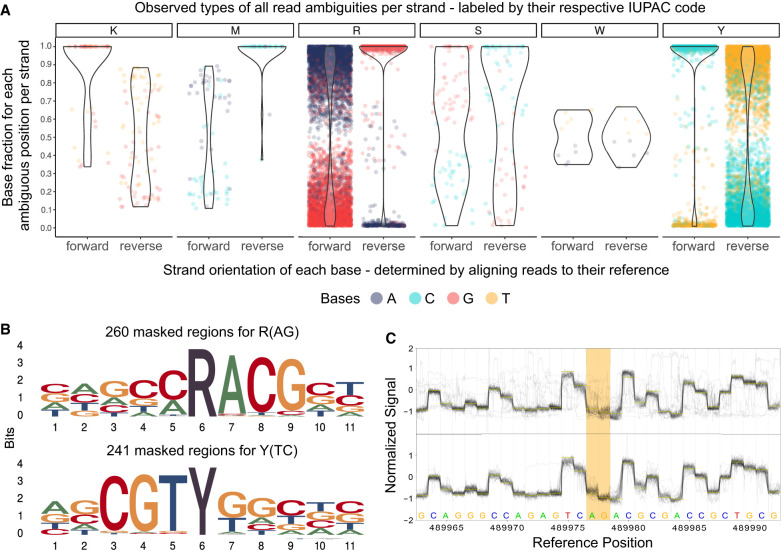
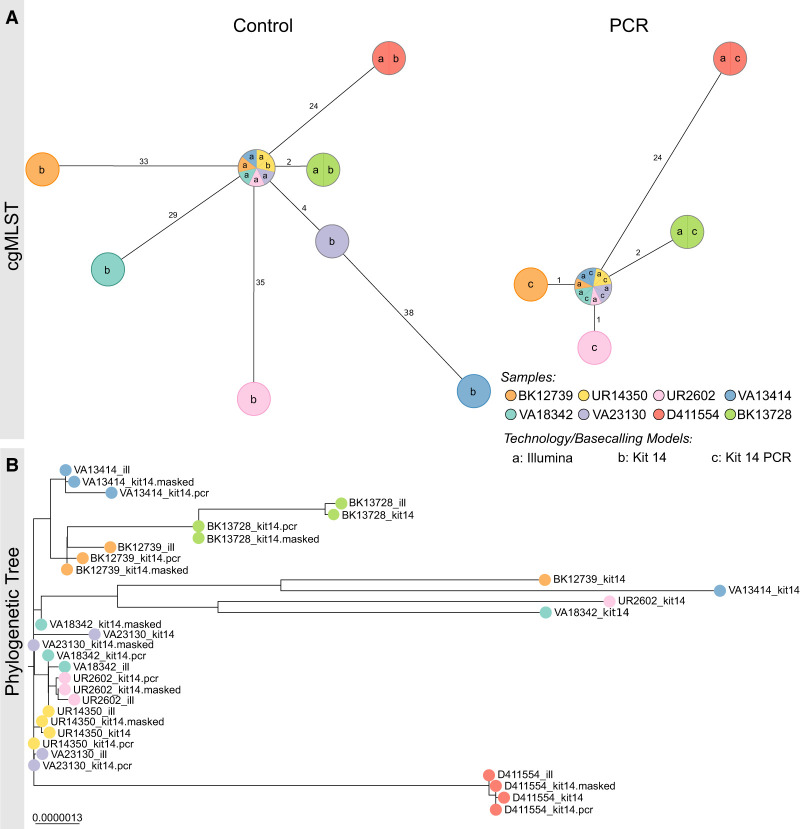
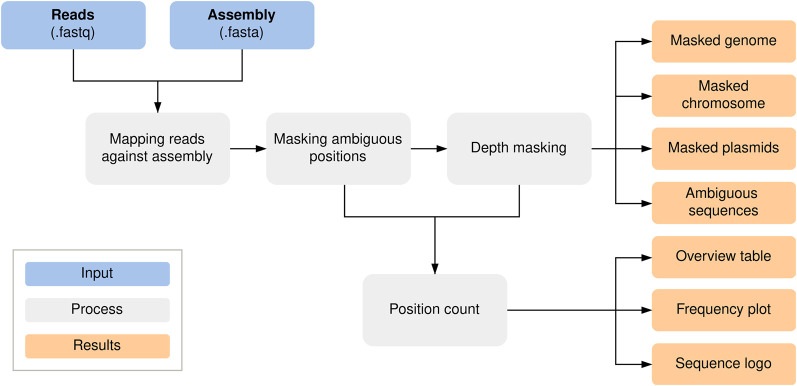
Our study investigates the effectiveness of Oxford Nanopore Technologies for accurate outbreak tracing by resequencing 33 isolates of a 3-year-long Klebsiella pneumoniae outbreak with Illumina short-read sequencing data as the point of reference. We detect considerable base errors through cgMLST and phylogenetic analysis of genomes sequenced with Oxford Nanopore Technologies, leading to the false exclusion of some outbreak-related strains from the outbreak cluster. Nearby methylation sites cause these errors and can also be found in other species besides K. pneumoniae Based on these data, we explore PCR-based sequencing and a masking strategy, which both successfully address these inaccuracies and ensure accurate outbreak tracing. We offer our masking strategy as a bioinformatic workflow (MPOA) to identify and mask problematic genome positions in a reference-free manner. Our research highlights limitations in using Oxford Nanopore Technologies for sequencing prokaryotic organisms, especially for investigating outbreaks. For time-critical projects that cannot wait for further technological developments by Oxford Nanopore Technologies, our study recommends either using PCR-based sequencing or using our provided bioinformatic workflow. We advise that read mapping-based quality control of genomes should be provided when publishing results.
© 2024 Lohde et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
Similar articles
-
Closing the gap: Oxford Nanopore Technologies R10 sequencing allows comparable results to Illumina sequencing for SNP-based outbreak investigation of bacterial pathogens.J Clin Microbiol. 2024 May 8;62(5):e0157623. doi: 10.1128/jcm.01576-23. Epub 2024 Mar 5. J Clin Microbiol. 2024. PMID: 38441926 Free PMC article.
-
Evaluation of the accuracy of bacterial genome reconstruction with Oxford Nanopore R10.4.1 long-read-only sequencing.Microb Genom. 2024 May;10(5):001246. doi: 10.1099/mgen.0.001246. Microb Genom. 2024. PMID: 38713194 Free PMC article.
-
The rapid detection of a neonatal unit outbreak of a wild-type Klebsiella variicola using decentralized Oxford Nanopore sequencing.Antimicrob Resist Infect Control. 2025 Feb 7;14(1):6. doi: 10.1186/s13756-025-01529-2. Antimicrob Resist Infect Control. 2025. PMID: 39920743 Free PMC article.
-
Evaluation of Illumina and Oxford Nanopore Sequencing for the Study of DNA Methylation in Alzheimer's Disease and Frontotemporal Dementia.Int J Mol Sci. 2025 Apr 28;26(9):4198. doi: 10.3390/ijms26094198. Int J Mol Sci. 2025. PMID: 40362435 Free PMC article. Review.
-
Applications of Nanopore sequencing in precision cancer medicine.Int J Cancer. 2024 Dec 15;155(12):2129-2140. doi: 10.1002/ijc.35100. Epub 2024 Jul 19. Int J Cancer. 2024. PMID: 39031959 Review.
Cited by
-
Decoding bacterial methylomes in four public health-relevant microbial species: nanopore sequencing enables reproducible analysis of DNA modifications.BMC Genomics. 2025 Apr 23;26(1):394. doi: 10.1186/s12864-025-11592-z. BMC Genomics. 2025. PMID: 40269718 Free PMC article.
-
High intra-laboratory reproducibility of nanopore sequencing in bacterial species underscores advances in its accuracy.Microb Genom. 2025 Mar;11(3):001372. doi: 10.1099/mgen.0.001372. Microb Genom. 2025. PMID: 40117330 Free PMC article.
-
Oxford Nanopore's 2024 sequencing technology for Listeria monocytogenes outbreak detection and source attribution: progress and clone-specific challenges.J Clin Microbiol. 2024 Nov 13;62(11):e0108324. doi: 10.1128/jcm.01083-24. Epub 2024 Oct 4. J Clin Microbiol. 2024. PMID: 39365069 Free PMC article.
-
Closing the gap: Oxford Nanopore Technologies R10 sequencing allows comparable results to Illumina sequencing for SNP-based outbreak investigation of bacterial pathogens.J Clin Microbiol. 2024 May 8;62(5):e0157623. doi: 10.1128/jcm.01576-23. Epub 2024 Mar 5. J Clin Microbiol. 2024. PMID: 38441926 Free PMC article.
-
Comparison of Illumina and Oxford Nanopore sequencing data quality for Clostridioides difficile genome analysis and their application for epidemiological surveillance.BMC Genomics. 2025 Jan 30;26(1):92. doi: 10.1186/s12864-025-11267-9. BMC Genomics. 2025. PMID: 39885402 Free PMC article.
References
-
- Abe R, Oyama F, Akeda Y, Nozaki M, Hatachi T, Okamoto Y, Yoshida H, Hamaguchi S, Tomono K, Matsumoto Y, et al. 2021. Hospital-wide outbreaks of carbapenem-resistant Enterobacteriaceae horizontally spread through a clonal plasmid harbouring blaIMP-1 in children's hospitals in Japan. J Antimicrob Chemother 76: 3314–3317. 10.1093/jac/dkab303 - DOI - PubMed
-
- Bialek-Davenet S, Criscuolo A, Ailloud F, Passet V, Jones L, Delannoy-Vieillard A-S, Garin B, Le Hello S, Arlet G, Nicolas-Chanoine M-H, et al. 2014. Genomic definition of hypervirulent and multidrug-resistant Klebsiella pneumoniae clonal groups. Emerg Infect Dis 20: 1812–1820. 10.3201/eid2011.140206 - DOI - PMC - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources