

Rifaximin prophylaxis causes resistance to the last-resort antibiotic daptomycin
- PMID: 39443798
- PMCID: PMC11602712
- DOI: 10.1038/s41586-024-08095-4
Rifaximin prophylaxis causes resistance to the last-resort antibiotic daptomycin
Abstract
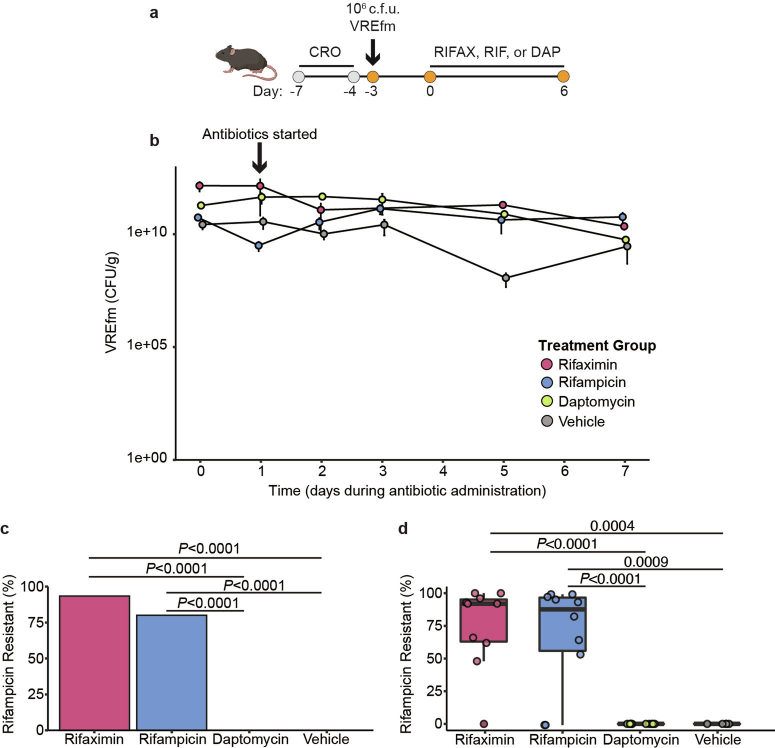
Multidrug-resistant bacterial pathogens like vancomycin-resistant Enterococcus faecium (VREfm) are a critical threat to human health1. Daptomycin is a last-resort antibiotic for VREfm infections with a novel mode of action2, but for which resistance has been widely reported but is unexplained. Here we show that rifaximin, an unrelated antibiotic used prophylactically to prevent hepatic encephalopathy in patients with liver disease3, causes cross-resistance to daptomycin in VREfm. Amino acid changes arising within the bacterial RNA polymerase in response to rifaximin exposure cause upregulation of a previously uncharacterized operon (prdRAB) that leads to cell membrane remodelling and cross-resistance to daptomycin through reduced binding of the antibiotic. VREfm with these mutations are spread globally, making this a major mechanism of resistance. Rifaximin has been considered 'low risk' for the development of antibiotic resistance. Our study shows that this assumption is flawed and that widespread rifaximin use, particularly in patients with liver cirrhosis, may be compromising the clinical use of daptomycin, a major last-resort intervention for multidrug-resistant pathogens. These findings demonstrate how unanticipated antibiotic cross-resistance can undermine global strategies designed to preserve the clinical use of critical antibiotics.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures













References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
