

Epigenome-wide analysis across the development span of pediatric acute lymphoblastic leukemia: backtracking to birth
- PMID: 39443995
- PMCID: PMC11515509
- DOI: 10.1186/s12943-024-02118-4
Epigenome-wide analysis across the development span of pediatric acute lymphoblastic leukemia: backtracking to birth
Abstract
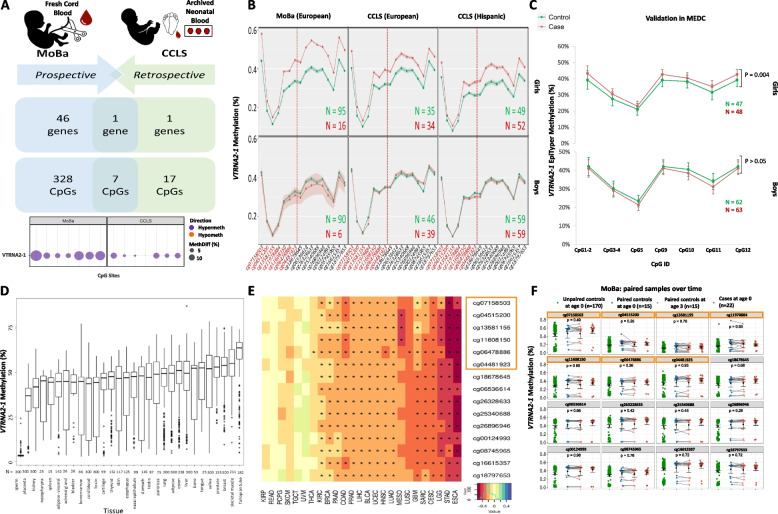
Background: Cancer is the leading cause of disease-related mortality in children. Causes of leukemia, the most common form, are largely unknown. Growing evidence points to an origin in-utero, when global redistribution of DNA methylation occurs driving tissue differentiation.
Methods: Epigenome-wide DNA methylation was profiled in surrogate (blood) and target (bone marrow) tissues at birth, diagnosis, remission and relapse of pediatric pre-B acute lymphoblastic leukemia (pre-B ALL) patients. Double-blinded analyses was performed between prospective cohorts extending from birth to diagnosis and retrospective studies backtracking from clinical disease to birth. Validation was carried out using independent technologies and populations.
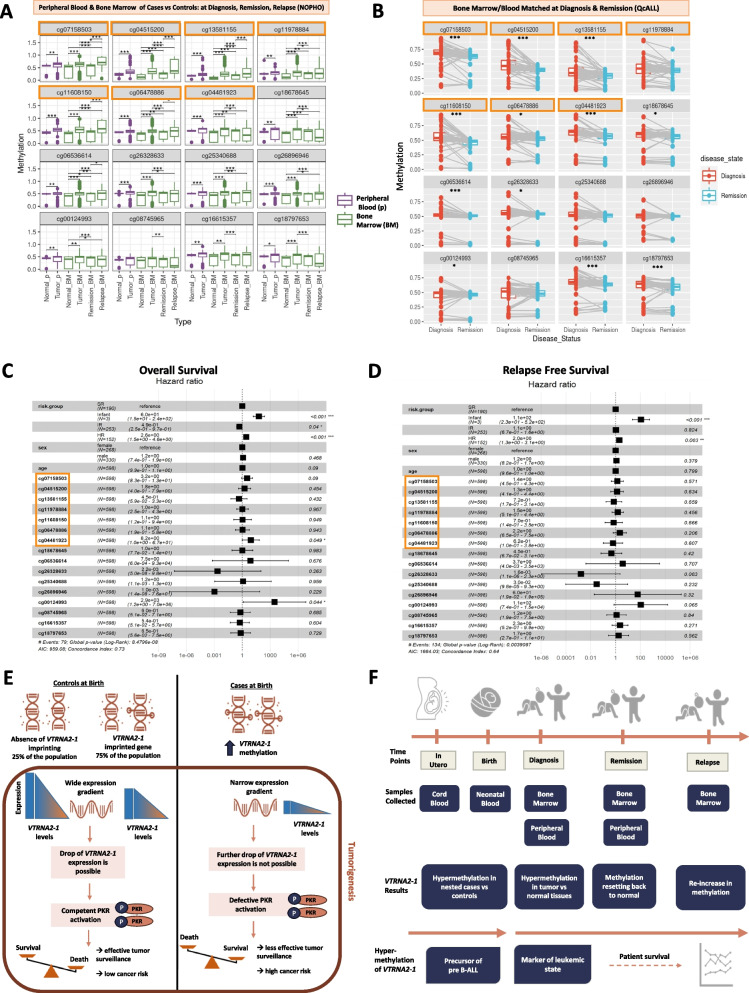
Results: The imprinted and immuno-modulating VTRNA2-1 was hypermethylated (FDR<0.05) at birth in nested cases relative to controls in all tested populations (totaling 317 cases and 483 controls), including European and Hispanic ancestries. VTRNA2-1 methylation was stable over follow-up years after birth and across surrogate, target and other tissues (n=5,023 tissues; 30 types). When profiled in leukemic tissues from two clinical cohorts (totaling 644 cases), VTRNA2-1 methylation exhibited higher levels at diagnosis relative to controls, it reset back to normal levels at remission, and then re-increased to above control levels at relapse. Hypermethylation was significantly associated with worse pre-B ALL patient survival and with reduced VTRNA2-1 expression (n=2,294 tissues; 26 types), supporting a functional and translational role for VTRNA2-1 methylation.
Conclusion: This study provides proof-of-concept to detect at birth epigenetic precursors of pediatric pre-B ALL. These alterations were reproducible with different technologies, in three continents and in two ethnicities, and can offer biomarkers for early detection and prognosis as well as actionable targets for therapy.
Keywords: VTRNA2-1; Birth cohort; DNA methylation; Epigenetics; Neonatal blood spots; Pediatric leukemia.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Erdmann F, Ghantous A, Schüz J. Environmental agents and childhood cancer. In: Nriagu J, editor. Encyclopedia of Environmental Health. 2nd ed. Burlington: Elsevier B.V.; 2019. p. 336–47. https://scholar.google.com/scholar_lookup?title=Encyclopedia+of+Environm....
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
