

Structure-Based Probe Reveals the Presence of Large Transthyretin Aggregates in Plasma of ATTR Amyloidosis Patients
- PMID: 39444930
- PMCID: PMC11494390
- DOI: 10.1016/j.jacbts.2024.05.013
Structure-Based Probe Reveals the Presence of Large Transthyretin Aggregates in Plasma of ATTR Amyloidosis Patients
Abstract
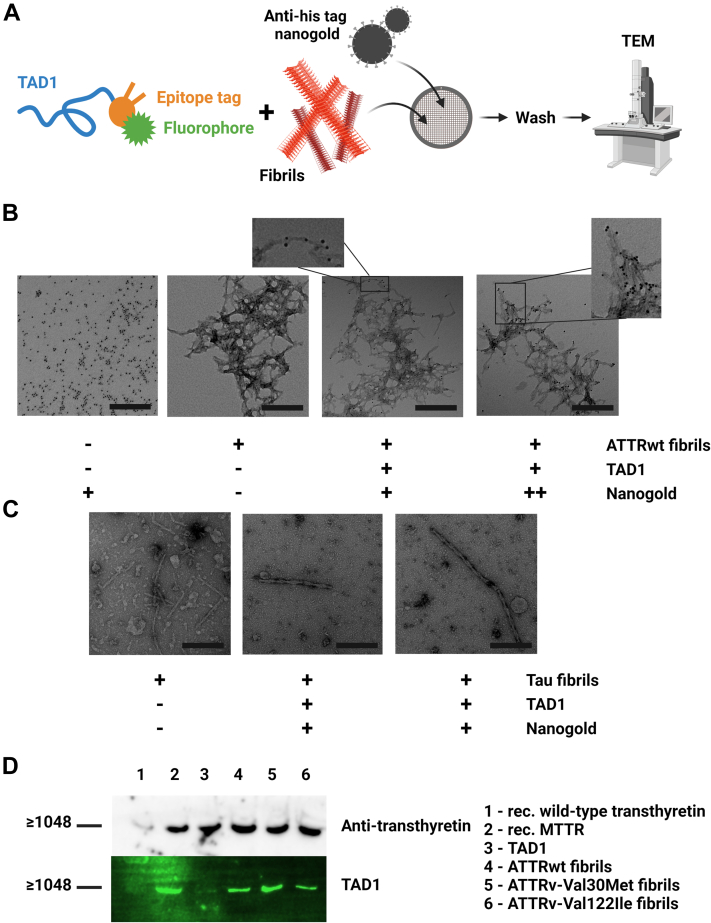
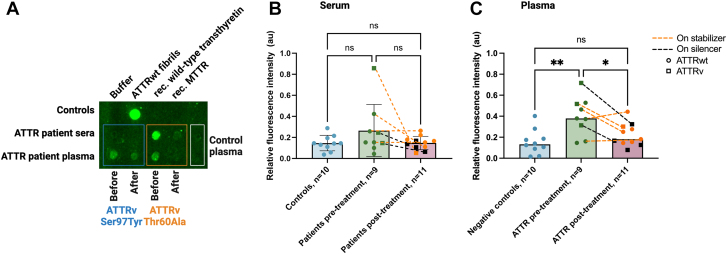
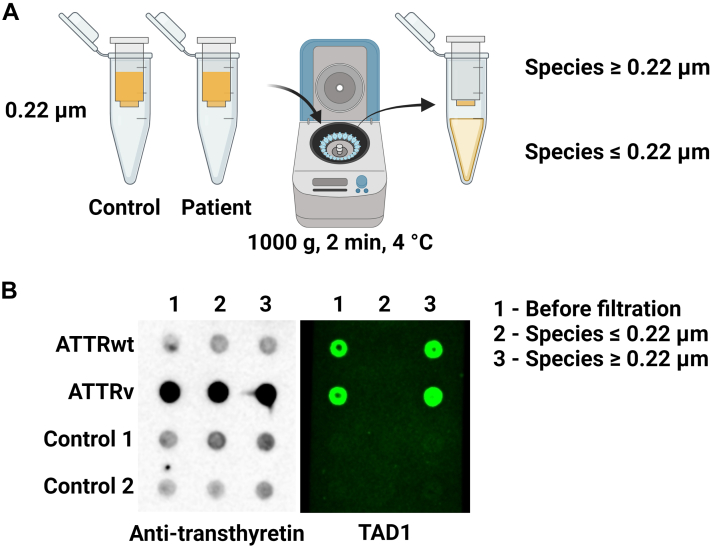
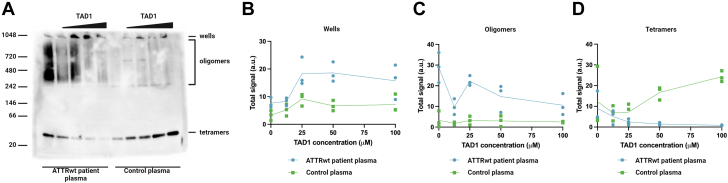
Amyloidogenic transthyretin (ATTR) amyloidosis is a relentlessly progressive disease caused by the misfolding and systemic accumulation of amyloidogenic transthyretin into amyloid fibrils. These fibrils cause diverse clinical phenotypes, mainly cardiomyopathy and/or polyneuropathy. Little is known about the aggregation of transthyretin during disease development and whether this has implications for diagnosis and treatment. Using the cryogenic electron microscopy structures of mature ATTR fibrils, we developed a peptide probe for fibril detection. With this probe, we have identified previously unknown aggregated transthyretin species in plasma of patients with ATTR amyloidosis. These species are large, non-native, and distinct from monomeric and tetrameric transthyretin. Observations from our study open many questions about the biology of ATTR amyloidosis and reveal a potential diagnostic and therapeutic target.
Keywords: amyloid; amyloidogenic transthyretin; biomarker; transthyretin.
© 2024 The Authors.
Conflict of interest statement
This work was supported by the American Heart Association Career Development Award (847236) (to Dr Saelices), the National Institutes of Health (NIH) Director's New Innovator Award (DP2-HL163810-01) (to Dr Saelices), the Welch Foundation Research Award (I-2121-20220331) (to Dr Saelices), the NIH R01 (R01-HL160892) (to Dr Grodin), and the NIH grant 1S10OD021685-01A1 received by the Electron Microscopy Core of UTSW. R. Pedretti and Dr Saelices are inventors on a patent application (Provisional Patent Application 63/352,521) submitted by the University of Texas Southwestern Medical Center that covers the composition and structure-based diagnostic methods related to cardiac ATTR amyloidosis. Dr Grodin has received honoraria for scientific consulting from Alnylam, Eidos/BridgeBio, Intellia, Pfizer, Alexion, AstraZeneca, and Tenax Therapeutics; and has received research funding from Pfizer, Eidos/BridgeBio, the Texas Health Resources Clinical Scholars fund, and the National Heart, Lung, and Blood Institute. Dr Masri has received research funding from Pfizer, Ionis/Akcea, Attralus, and Cytokinetics; and has received consulting fees from Cytokinetics, Bristol Myers Squibb, Eidos, Pfizer, Ionis, Lexicon, Alnylam, Attralus, Haya, Intellia, BioMarin, and Tenaya. Dr Saelices has received honoraria for scientific consulting for Intellia and Attralus; and has served as a member of the Advisory Board of Alexion. All other authors have reported they have no relationships relevant to the contents of this paper to disclose.PerspectivesCOMPETENCY IN MEDICAL KNOWLEDGE: ATTR amyloidosis is an under-recognized cause of heart failure due to lack of tools that can effectively distinguish between native and amyloidogenic transthyretin. Using structures of mature ATTR fibrils derived from patient tissue, we have developed a novel detection probe that addresses this unmet need. This probe not only binds ATTR fibrils with high affinity, but also large ATTR aggregates in plasma of patients with ATTR amyloidosis. TRANSLATIONAL OUTLOOK: Combined with previous results, our data unveils a novel biomarker for cardiac ATTR amyloidosis that has the potential to be exploited as a novel diagnostic and/or therapeutic target. Our results may be leveraged to develop a screening tool using blood samples to identify disease onset earlier than the current diagnostic standard. This will enable timely treatment, improve patient prognosis, and reduce burden on the health care system.
Figures


References
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
