

Epstein-Barr virus reactivation induces divergent abortive, reprogrammed, and host shutoff states by lytic progression
- PMID: 39446925
- PMCID: PMC11563402
- DOI: 10.1371/journal.ppat.1012341
Epstein-Barr virus reactivation induces divergent abortive, reprogrammed, and host shutoff states by lytic progression
Abstract
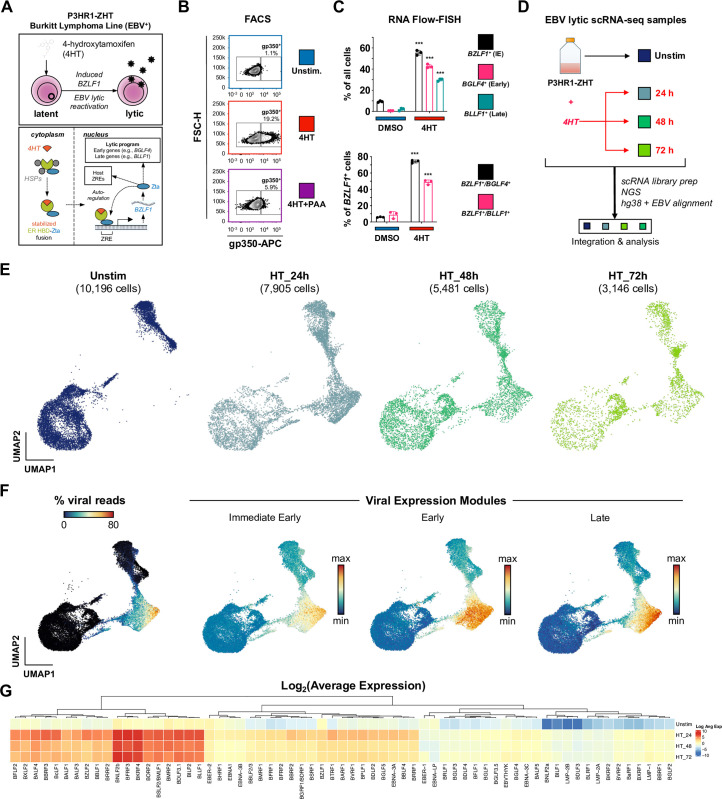
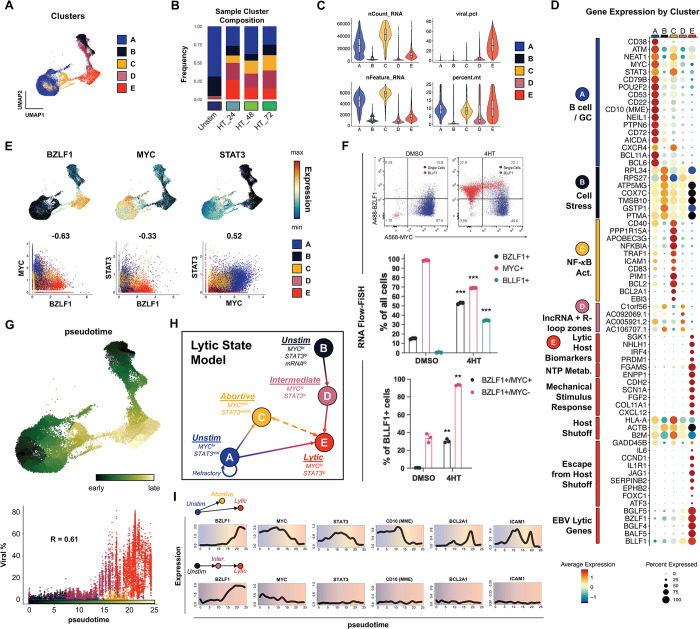
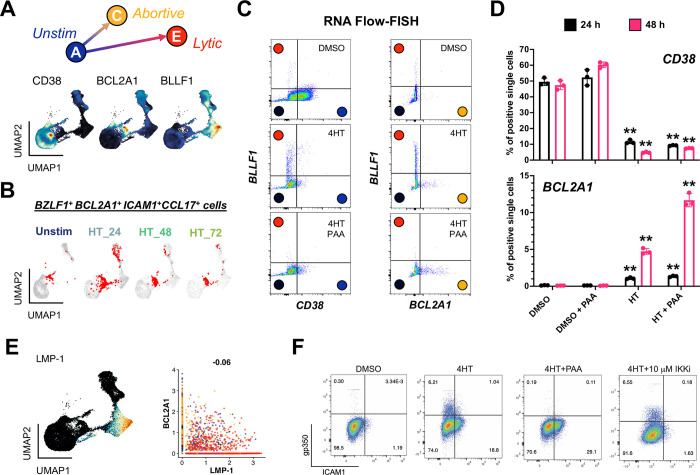
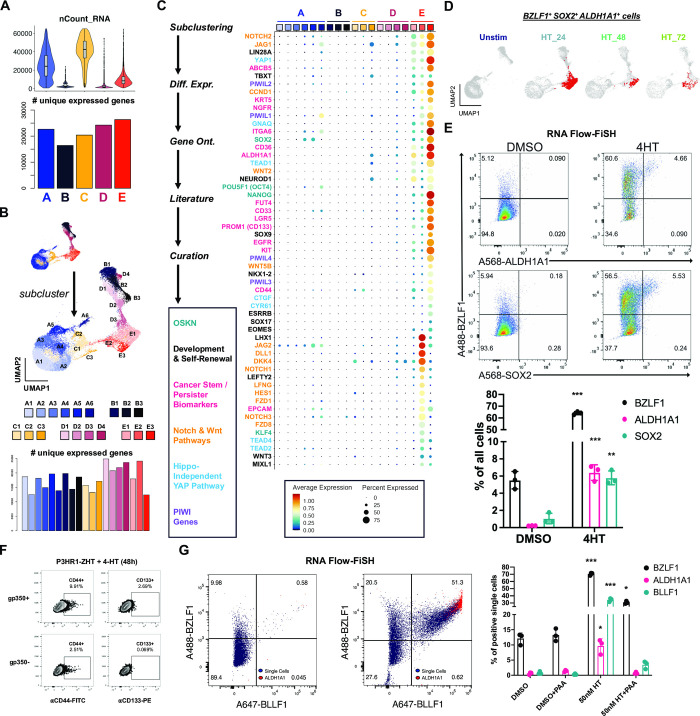
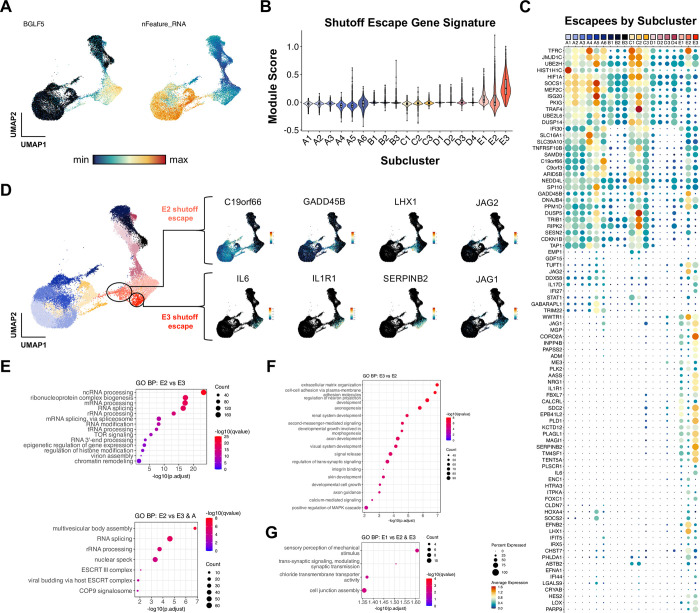
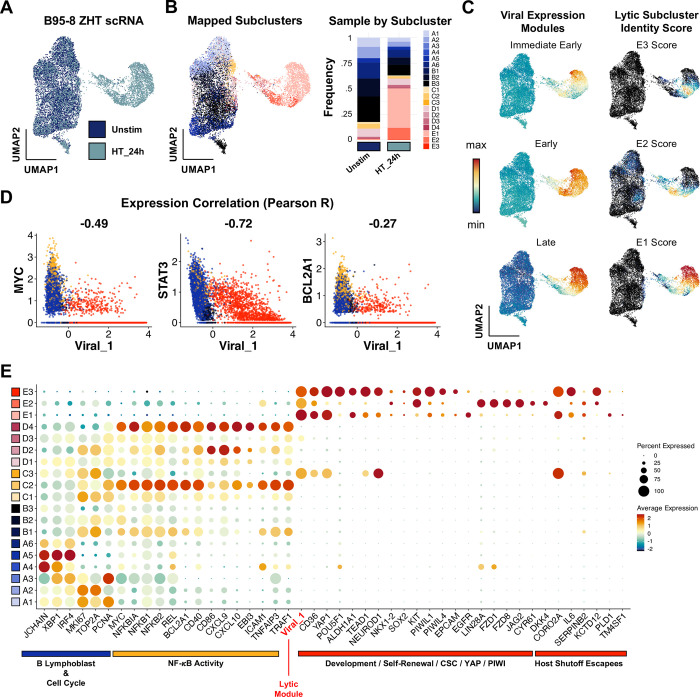
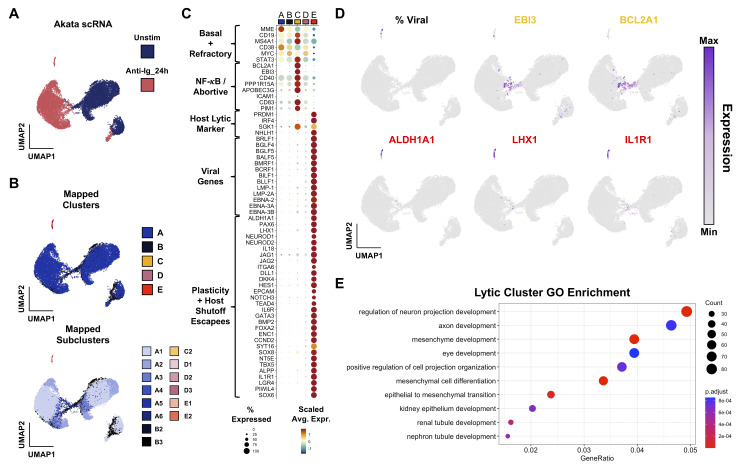
Viral infection leads to heterogeneous cellular outcomes ranging from refractory to abortive and fully productive states. Single cell transcriptomics enables a high resolution view of these distinct post-infection states. Here, we have interrogated the host-pathogen dynamics following reactivation of Epstein-Barr virus (EBV). While benign in most people, EBV is responsible for infectious mononucleosis, up to 2% of human cancers, and is a trigger for the development of multiple sclerosis. Following latency establishment in B cells, EBV reactivates and is shed in saliva to enable infection of new hosts. Beyond its importance for transmission, the lytic cycle is also implicated in EBV-associated oncogenesis. Conversely, induction of lytic reactivation in latent EBV-positive tumors presents a novel therapeutic opportunity. Therefore, defining the dynamics and heterogeneity of EBV lytic reactivation is a high priority to better understand pathogenesis and therapeutic potential. In this study, we applied single-cell techniques to analyze diverse fate trajectories during lytic reactivation in three B cell models. Consistent with prior work, we find that cell cycle and MYC expression correlate with cells refractory to lytic reactivation. We further found that lytic induction yields a continuum from abortive to complete reactivation. Abortive lytic cells upregulate NFκB and IRF3 pathway target genes, while cells that proceed through the full lytic cycle exhibit unexpected expression of genes associated with cellular reprogramming. Distinct subpopulations of lytic cells further displayed variable profiles for transcripts known to escape virus-mediated host shutoff. These data reveal previously unknown and promiscuous outcomes of lytic reactivation with broad implications for viral replication and EBV-associated oncogenesis.
Copyright: © 2024 SoRelle et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Update of
-
Epstein-Barr virus reactivation induces divergent abortive, reprogrammed, and host shutoff states by lytic progression.bioRxiv [Preprint]. 2024 Jun 14:2024.06.14.598975. doi: 10.1101/2024.06.14.598975. bioRxiv. 2024. Update in: PLoS Pathog. 2024 Oct 24;20(10):e1012341. doi: 10.1371/journal.ppat.1012341. PMID: 38915538 Free PMC article. Updated. Preprint.
Similar articles
-
Epstein-Barr virus reactivation induces divergent abortive, reprogrammed, and host shutoff states by lytic progression.bioRxiv [Preprint]. 2024 Jun 14:2024.06.14.598975. doi: 10.1101/2024.06.14.598975. bioRxiv. 2024. Update in: PLoS Pathog. 2024 Oct 24;20(10):e1012341. doi: 10.1371/journal.ppat.1012341. PMID: 38915538 Free PMC article. Updated. Preprint.
-
IRF6 controls Epstein-Barr virus (EBV) lytic reactivation and differentiation in EBV-infected epithelial cells.PLoS Pathog. 2025 Jun 26;21(6):e1013236. doi: 10.1371/journal.ppat.1013236. eCollection 2025 Jun. PLoS Pathog. 2025. PMID: 40569988 Free PMC article.
-
Assessing the fitness of Epstein-Barr virus following its reactivation.J Virol. 2025 Jul 22;99(7):e0062625. doi: 10.1128/jvi.00626-25. Epub 2025 May 30. J Virol. 2025. PMID: 40444990 Free PMC article.
-
EBV Reactivation from Latency Is a Degrading Experience for the Host.Viruses. 2023 Mar 11;15(3):726. doi: 10.3390/v15030726. Viruses. 2023. PMID: 36992435 Free PMC article. Review.
-
The PD-1/PD-L1 pathway and Epstein-Barr virus.Eur J Med Res. 2025 Jun 18;30(1):486. doi: 10.1186/s40001-025-02694-1. Eur J Med Res. 2025. PMID: 40533842 Free PMC article. Review.
Cited by
-
Fatty acid desaturases link cell metabolism pathways to promote proliferation of Epstein-Barr virus-infected B cells.PLoS Pathog. 2025 May 22;21(5):e1012685. doi: 10.1371/journal.ppat.1012685. eCollection 2025 May. PLoS Pathog. 2025. PMID: 40403013 Free PMC article.
-
EBV Reactivation-associated gene signature predicts poor prognosis in nasopharyngeal carcinoma.J Transl Med. 2025 Jun 2;23(1):616. doi: 10.1186/s12967-025-06549-5. J Transl Med. 2025. PMID: 40457434 Free PMC article.
-
mSphere of Influence: The integral art of resolving host-virus interactions.mSphere. 2025 Apr 29;10(4):e0104024. doi: 10.1128/msphere.01040-24. Epub 2025 Mar 20. mSphere. 2025. PMID: 40111039 Free PMC article.
-
E2F1 suppresses Epstein-Barr virus lytic reactivation through cellular and viral transcriptional networks.PLoS Pathog. 2025 Aug 7;21(8):e1013410. doi: 10.1371/journal.ppat.1013410. eCollection 2025 Aug. PLoS Pathog. 2025. PMID: 40773523 Free PMC article.
-
Epstein-Barr virus hijacks histone demethylase machinery to drive epithelial malignancy progression through KDM5B upregulation.Signal Transduct Target Ther. 2025 Mar 10;10(1):83. doi: 10.1038/s41392-025-02163-5. Signal Transduct Target Ther. 2025. PMID: 40059116 Free PMC article.
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
