

Stepwise phosphorylation and SUMOylation of PIDD1 drive PIDDosome assembly in response to DNA repair failure
- PMID: 39448602
- PMCID: PMC11502896
- DOI: 10.1038/s41467-024-53412-0
Stepwise phosphorylation and SUMOylation of PIDD1 drive PIDDosome assembly in response to DNA repair failure
Abstract
SUMOylation regulates numerous cellular stress responses, yet targets in the apoptotic machinery remain elusive. We show that a single, DNA damage-induced monoSUMOylation event controls PIDDosome (PIDD1/RAIDD/caspase-2) formation and apoptotic death in response to unresolved DNA interstrand crosslinks (ICLs). SUMO-1 conjugation occurs on conserved K879 in the PIDD1 death domain (DD); is catalyzed by PIAS1 and countered by SENP3; and is triggered by ATR phosphorylation of neighboring T788 in the PIDD1 DD, which enables PIAS1 docking. Phospho/SUMO-PIDD1 proteins are captured by nucleolar RAIDD monomers via a SUMO-interacting motif (SIM) in the RAIDD DD, thus compartmentalizing nascent PIDDosomes for caspase-2 recruitment. Denying SUMOylation or the SUMO-SIM interaction spares the onset of PIDDosome assembly but blocks its completion, thus eliminating the apoptotic response to ICL repair failure. Conversely, removal of SENP3 forces apoptosis, even in cells with tolerable ICL levels. SUMO-mediated PIDDosome control is also seen in response to DNA breaks but not supernumerary centrosomes. These results illuminate PIDDosome formation in space and time and identify a direct role for SUMOylation in the assembly of a major pro-apoptotic device.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
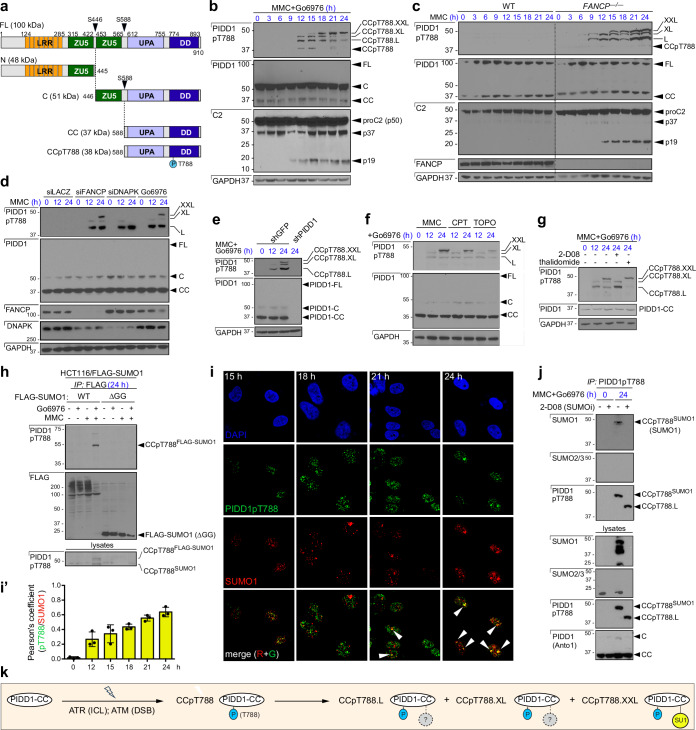
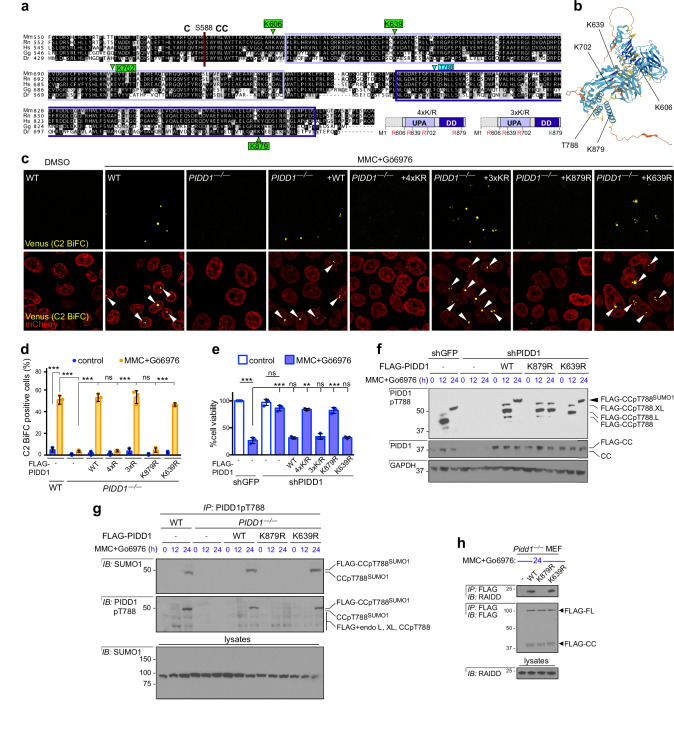
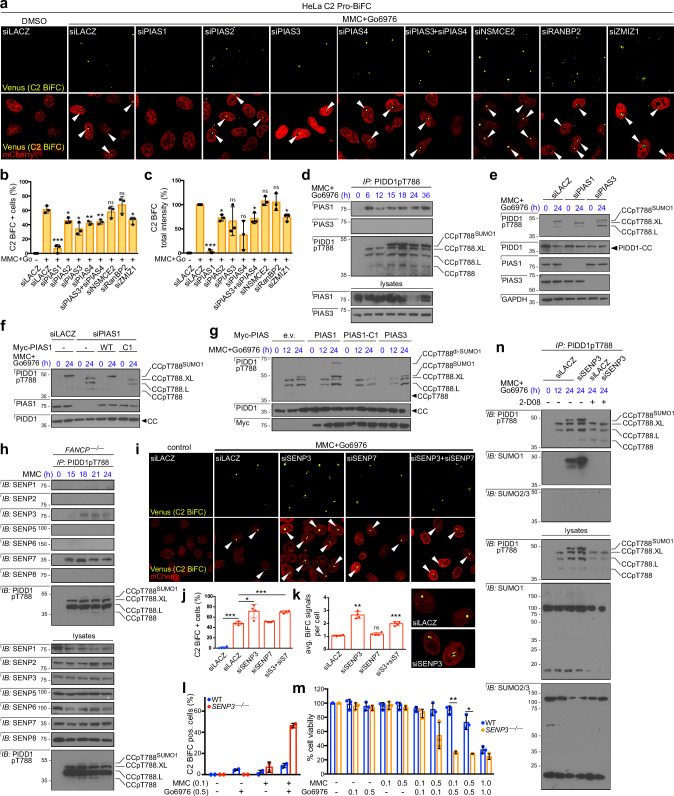
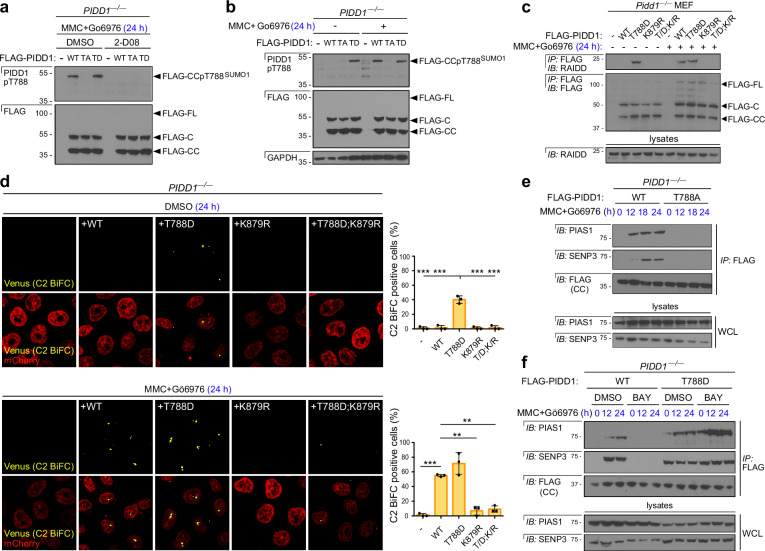
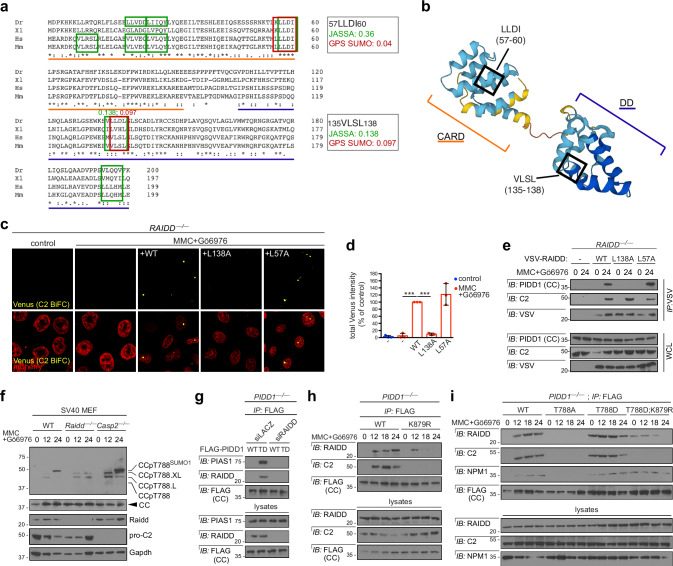
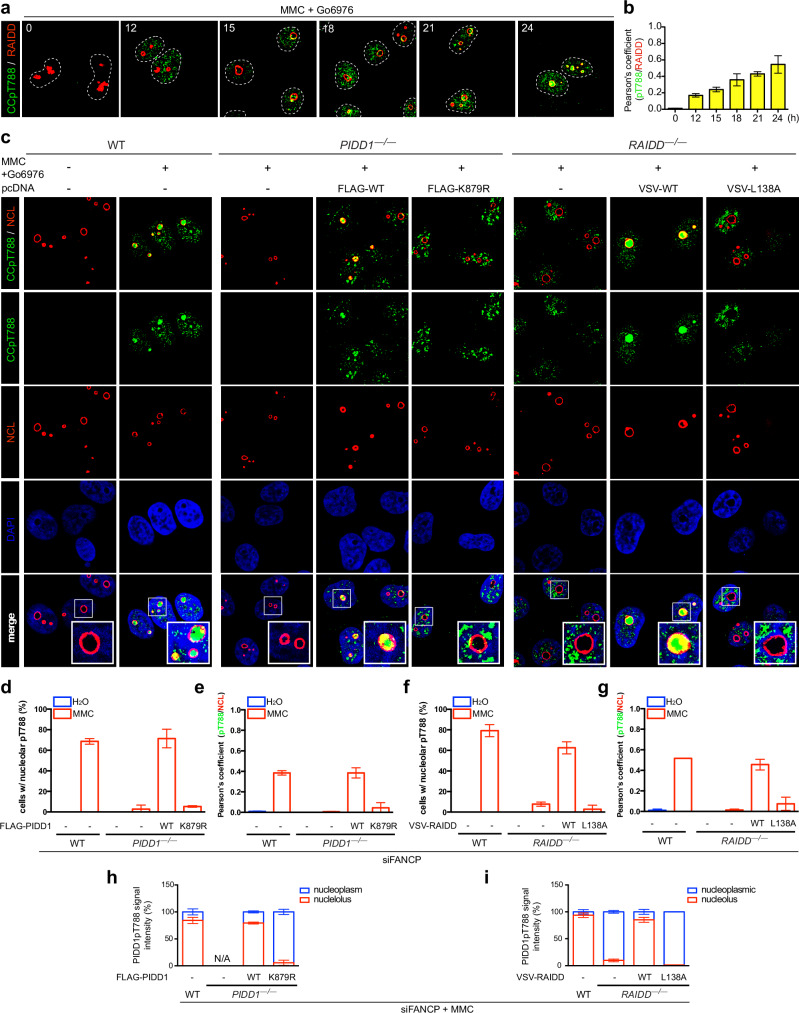
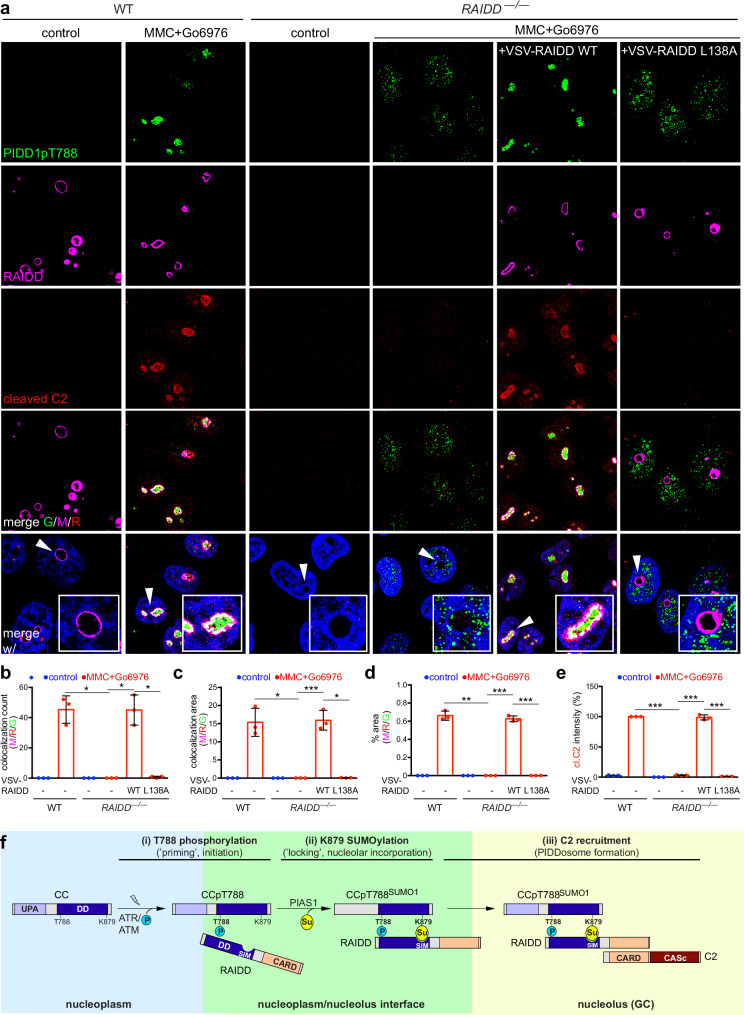
References
-
- Tinel, A. & Tschopp, J. The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science304, 843–846 (2004). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 CA178162/CA/NCI NIH HHS/United States
- R01CA178162/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- RO1GM135301/U.S. Department of Health & Human Services | NIH | National Institute of General Medical Sciences (NIGMS)
- R01 GM135301/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Miscellaneous
