

Emergence of ECSA-IOL E1-K211E/E2-V264A Lineage of Chikungunya virus during Malaysian 2021 outbreak
- PMID: 39448916
- PMCID: PMC11515639
- DOI: 10.1186/s12879-024-10102-y
Emergence of ECSA-IOL E1-K211E/E2-V264A Lineage of Chikungunya virus during Malaysian 2021 outbreak
Abstract
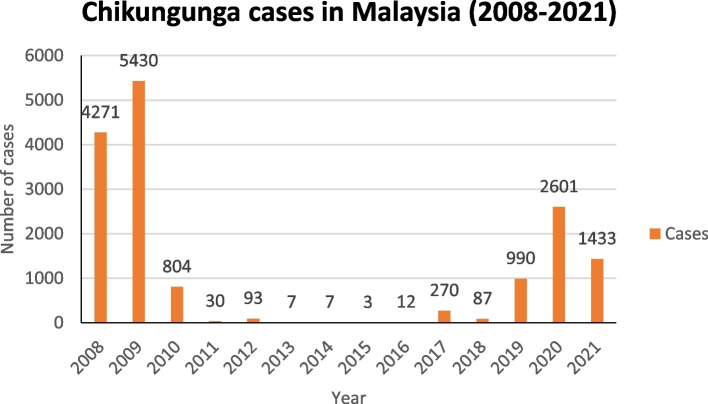
Background: Chikungunya cases was reported to be on the rise in Malaysia from 2019 to 2021. Although potential endemicity was described previously, genotype shift during 2008 outbreak originating from the 2004 Indian Ocean Islands outbreak presents the probability of current CHIKV spread from neighboring countries. This is due to the prevalence of the new IOL sub-lineage which consists of E1-226A wildtype or reverted strains that are circulating in the Indian subcontinent before spreading to neighboring Thailand during 2018-2019 outbreak.
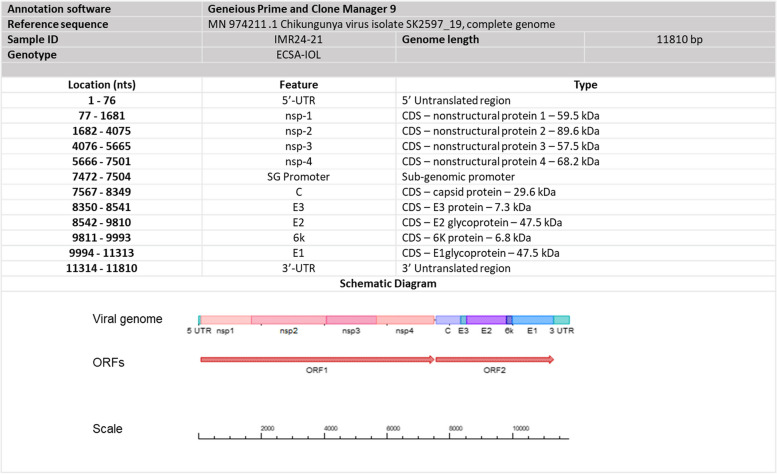
Method: In this study, samples received mostly from the Tangkak, Johor were analyzed. A total 56 CHIKV positive serum samples received in 2021 by Institute of Medical Research Malaysia (IMR), were collected based on sample selection criteria. Selected samples were subjected to total RNA extraction, whole-genome sequencing as well as bioinformatic analysis such as phylogenetic, variant and mutation analysis.
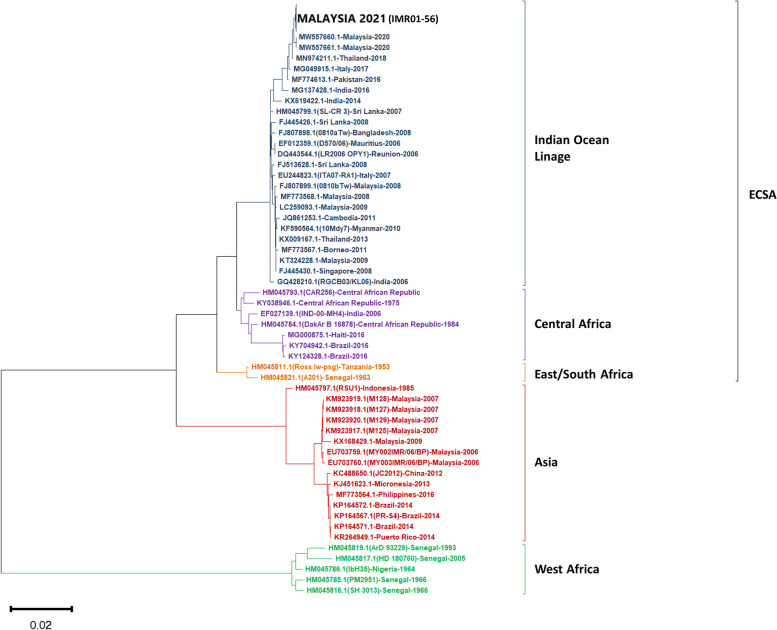
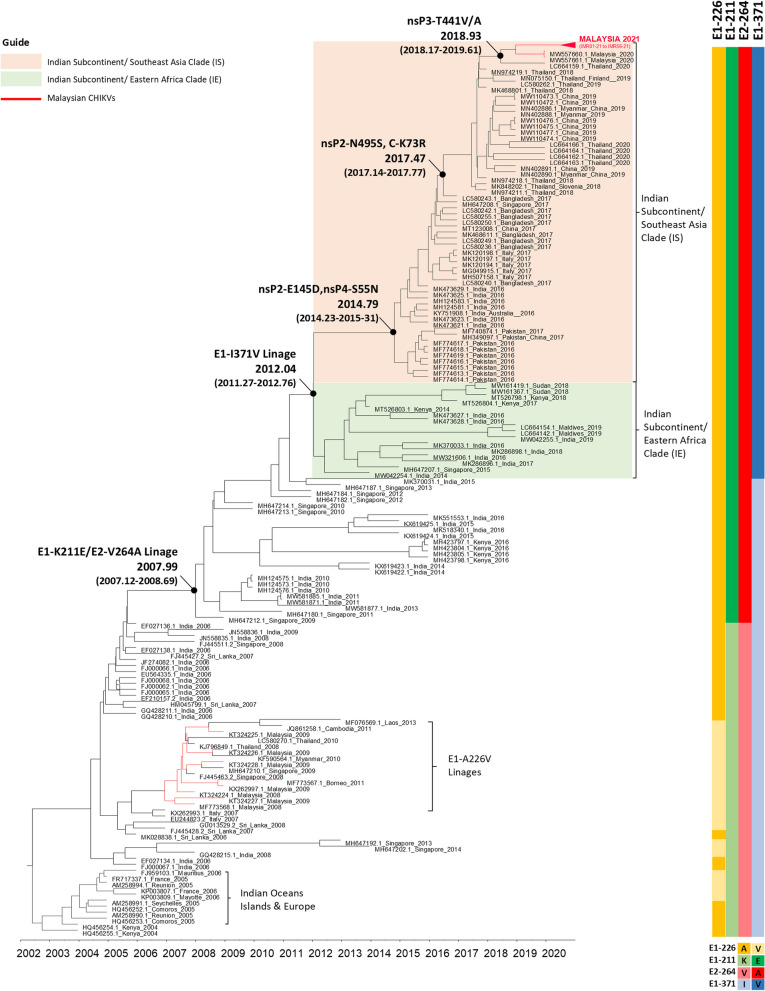
Results: Based on the genomic and phylogenetic analysis, the CHIKV samples from 2021 outbreak were of ECSA-IOL genotype. Genome similarity analysis also revealed that these CHIKVs were highly similar to 2018-2019 outbreak strain from Thailand. In comparison to the 2008 outbreak CHIKV isolate, the current CHIKVs lacked the E1-A226V mutation and harbored the new E1-K211E/E2-V264A sub-linage mutation. Since the E1-K211E/E2-V264A mutation facilitates adaptation to Ae. aegypti as opposed to the E1-A226V mutation which improves adaptation to Ae. albopictus, the emergence 2021 CHIKV outbreak in Malaysia can be postulated due to vector shift. Interestingly, a novel nsP3-T441A/V mutation detected in this study, may also play a role in virus transmission, pathogenicity, fitness and vector adaptation.
Conclusion: In summary, the current CHIKV outbreak are strains originated from the Indian subcontinent through Thailand which may have capitalized on vector shifting by adapting to Ae. aegypti. The presence of novel nsP3-T441A/V mutation may also contribute to the spread of this virus across peninsular Malaysia.
Keywords: CHIKV; Chikungunya; Malaysia; Sequencing; Whole-genome.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Chikungunya virus infection: molecular biology, clinical characteristics, and epidemiology in Asian countries.J Biomed Sci. 2021 Dec 2;28(1):84. doi: 10.1186/s12929-021-00778-8. J Biomed Sci. 2021. PMID: 34857000 Free PMC article. Review.
-
Emergence of a novel chikungunya virus strain bearing the E1:V80A substitution, out of the Mombasa, Kenya 2017-2018 outbreak.PLoS One. 2020 Nov 6;15(11):e0241754. doi: 10.1371/journal.pone.0241754. eCollection 2020. PLoS One. 2020. PMID: 33156857 Free PMC article.
-
Large-scale outbreak of Chikungunya virus infection in Thailand, 2018-2019.PLoS One. 2021 Mar 10;16(3):e0247314. doi: 10.1371/journal.pone.0247314. eCollection 2021. PLoS One. 2021. PMID: 33690657 Free PMC article.
-
A Novel Sub-Lineage of Chikungunya Virus East/Central/South African Genotype Indian Ocean Lineage Caused Sequential Outbreaks in Bangladesh and Thailand.Viruses. 2020 Nov 17;12(11):1319. doi: 10.3390/v12111319. Viruses. 2020. PMID: 33213040 Free PMC article.
-
Chikungunya: Its History in Africa and Asia and Its Spread to New Regions in 2013-2014.J Infect Dis. 2016 Dec 15;214(suppl 5):S436-S440. doi: 10.1093/infdis/jiw391. J Infect Dis. 2016. PMID: 27920169 Review.
Cited by
-
Prevalence of chikungunya virus infection in Sabah, Malaysia during 2017-2020.Trop Med Health. 2025 Apr 21;53(1):55. doi: 10.1186/s41182-025-00735-3. Trop Med Health. 2025. PMID: 40259363 Free PMC article.
-
The Re-emergence of Chikungunya in Sri Lanka: A Genomic investigation.medRxiv [Preprint]. 2025 May 23:2025.05.23.25328206. doi: 10.1101/2025.05.23.25328206. medRxiv. 2025. PMID: 40661282 Free PMC article. Preprint.
References
-
- Powers A. Chikungunya virus outbreak expansion and microevolutionary events affecting epidemiology and epidemic potential. Res Rep Trop Med. 2015;2015:6:11–19.
-
- Caglioti C, et al. Chikungunya virus infection: an overview. New Microbiol. 2013;36(3):211–27. - PubMed
-
- Kam YW, et al. Immuno-biology of Chikungunya and implications for disease intervention. Microbes Infect. 2009;11(14–15):1186–96. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
