

Genetic analysis using next-generation sequencing and multiplex ligation probe amplification in Chinese aniridia patients
- PMID: 39449022
- PMCID: PMC11515619
- DOI: 10.1186/s13023-024-03388-3
Genetic analysis using next-generation sequencing and multiplex ligation probe amplification in Chinese aniridia patients
Abstract
Background: Congenital aniridia is a rare pan-ocular disease characterized by complete irideremia, partial iridocoloboma. The progressive nature of aniridia is frequently accompanied by secondary ocular complications such as glaucoma and aniridia-associated keratopathy, which can lead to severely impaired vision or blindness. The genetic basis of aniridia has been the subject of numerous studies, leading to the development of innovative therapeutic options based on PAX6 nonsense mutations. Specific knowledge of the genetics of aniridia has become increasingly important. To report the clinical features, elucidate the genetic etiology, and reveal the mutational spectrum of congenital aniridia in the Chinese population, sixty patients with congenital aniridia from 51 families were recruited. Candidate genes associated with developmental eye diseases were identified and analyzed using panel-based next-generation sequencing (NGS), and mutations were confirmed through polymerase chain reaction and Sanger sequencing. Multiplex ligation probe amplification (MLPA) of PAX6 and FOXC1 was performed to detect copy number variations in the patients without intragenic mutations.
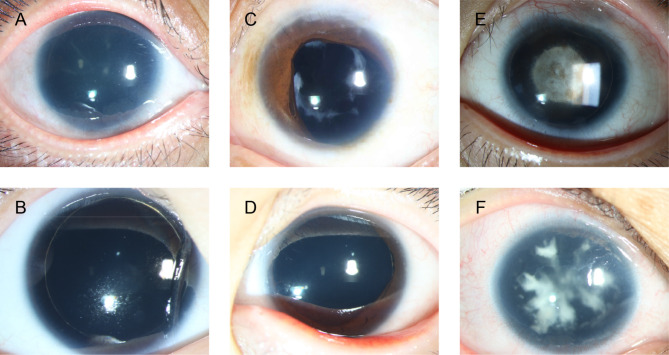
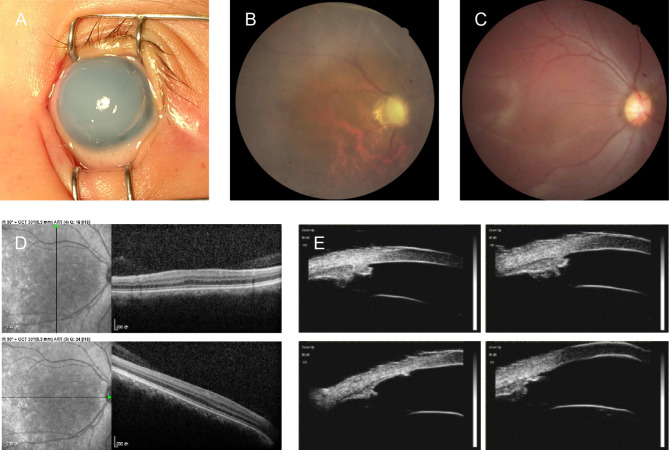
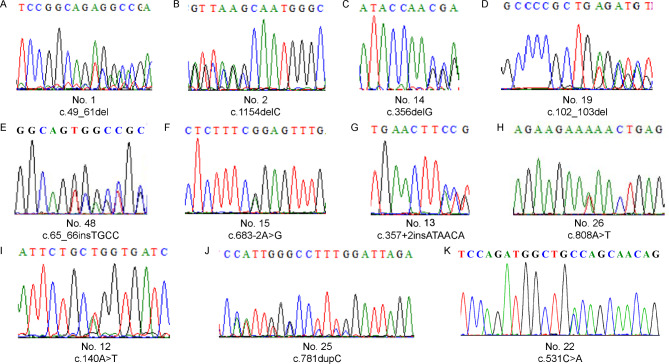
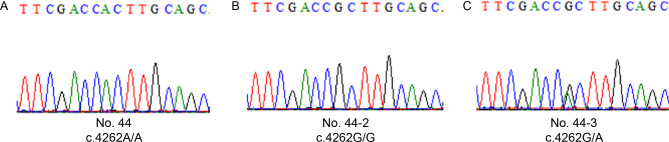
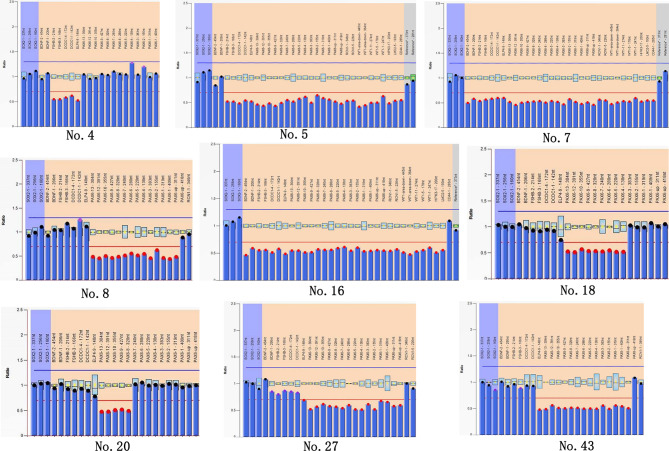
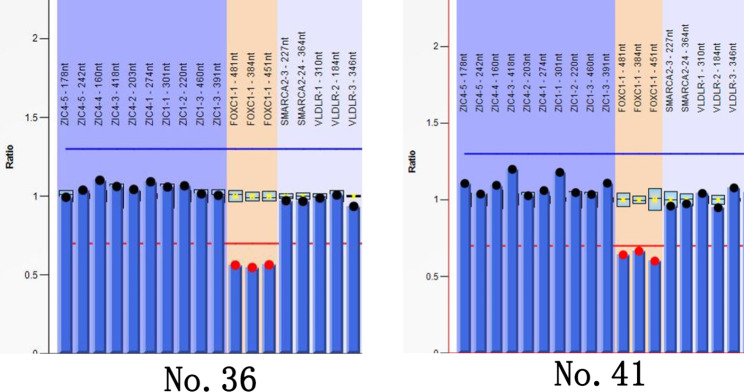
Results: Clinical examination revealed complete iris hypoplasia in 58 patients and partial iris hypoplasia in two patients. Additionally, two patients were diagnosed with Wilms' tumor-aniridia-genital anomalies-retardation syndrome and nephroblastoma. By combining panel-based NGS and MLPA, 43 intragenic mutations or deletions of PAX6, FOXC1, and BCOR were identified in 59 patients, including 33 point mutations (76.7%) in 43 patients and 10 deletions (23.3%) in 16 patients. The total detection rate was 98.3%. Phenotypic variation was observed between and within families.
Conclusions: Variations in PAX6 and its adjacent regions were the predominant causes of aniridia in China. In addition to intragenic point mutations in PAX6, deletion of PAX6 or its adjacent genes is a common cause of congenital aniridia. Furthermore, FOXC1 is an important gene associated with congenital aniridia. The combination of panel-based NGS and MLPA significantly enhanced the detection rate of gene mutations in patients with congenital aniridia.
Keywords: BCOR; FOXC1; PAX6; Congenital aniridia; Multiplex ligation probe amplification; Panel-based next-generation sequencing.
© 2024. The Author(s).
Conflict of interest statement
There are no conflicts of interest for any of the authors in this study.
Figures
Similar articles
-
Mutation spectrum of PAX6 and clinical findings in 95 Chinese patients with aniridia.Mol Vis. 2020 Mar 26;26:226-234. eCollection 2020. Mol Vis. 2020. PMID: 32214788 Free PMC article.
-
Large novel deletions detected in Chinese families with aniridia: correlation between genotype and phenotype.Mol Vis. 2011 Feb 19;17:548-57. Mol Vis. 2011. PMID: 21364908 Free PMC article.
-
Comparison between aniridia with and without PAX6 mutations: clinical and molecular analysis in 14 Korean patients with aniridia.Ophthalmology. 2012 Jun;119(6):1258-64. doi: 10.1016/j.ophtha.2011.12.010. Epub 2012 Feb 22. Ophthalmology. 2012. PMID: 22361317
-
Genetics and epidemiology of aniridia: Updated guidelines for genetic study.Arch Soc Esp Oftalmol (Engl Ed). 2021 Nov;96 Suppl 1:4-14. doi: 10.1016/j.oftale.2021.02.002. Epub 2021 Oct 22. Arch Soc Esp Oftalmol (Engl Ed). 2021. PMID: 34836588 Review.
-
Clinical and molecular aspects of congenital aniridia - A review of current concepts.Indian J Ophthalmol. 2022 Jul;70(7):2280-2292. doi: 10.4103/ijo.IJO_2255_21. Indian J Ophthalmol. 2022. PMID: 35791108 Free PMC article. Review.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
