

A single-stranded based library preparation method for virome characterization
- PMID: 39449043
- PMCID: PMC11515303
- DOI: 10.1186/s40168-024-01935-5
A single-stranded based library preparation method for virome characterization
Abstract
Background: The gut virome is an integral component of the gut microbiome, playing a crucial role in maintaining gut health. However, accurately depicting the entire gut virome is challenging due to the inherent diversity of genome types (dsDNA, ssDNA, dsRNA, and ssRNA) and topologies (linear, circular, or fragments), with subsequently biases associated with current sequencing library preparation methods. To overcome these problems and improve reproducibility and comparability across studies, universal or standardized virome sequencing library construction methods are highly needed in the gut virome study.
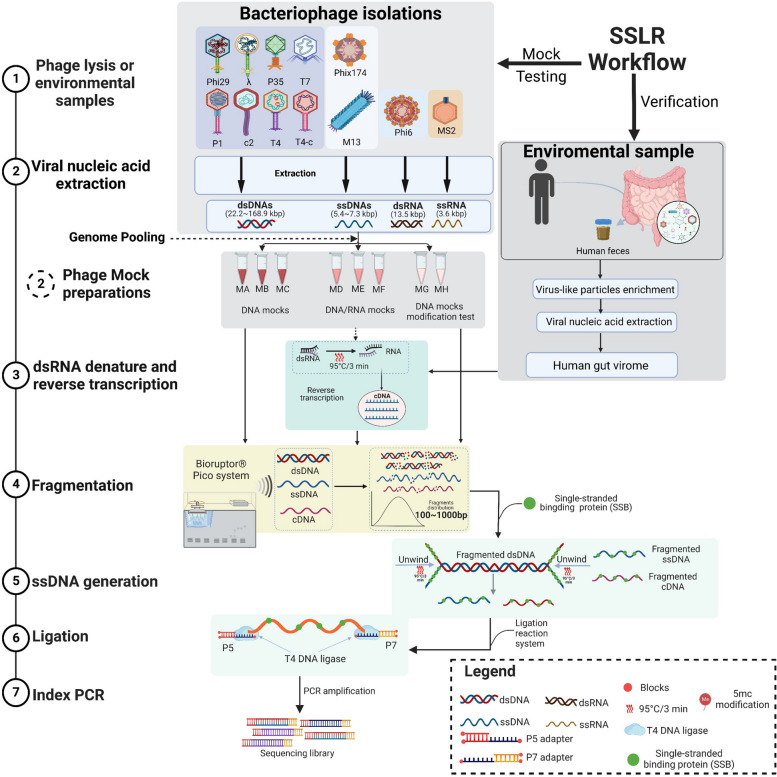
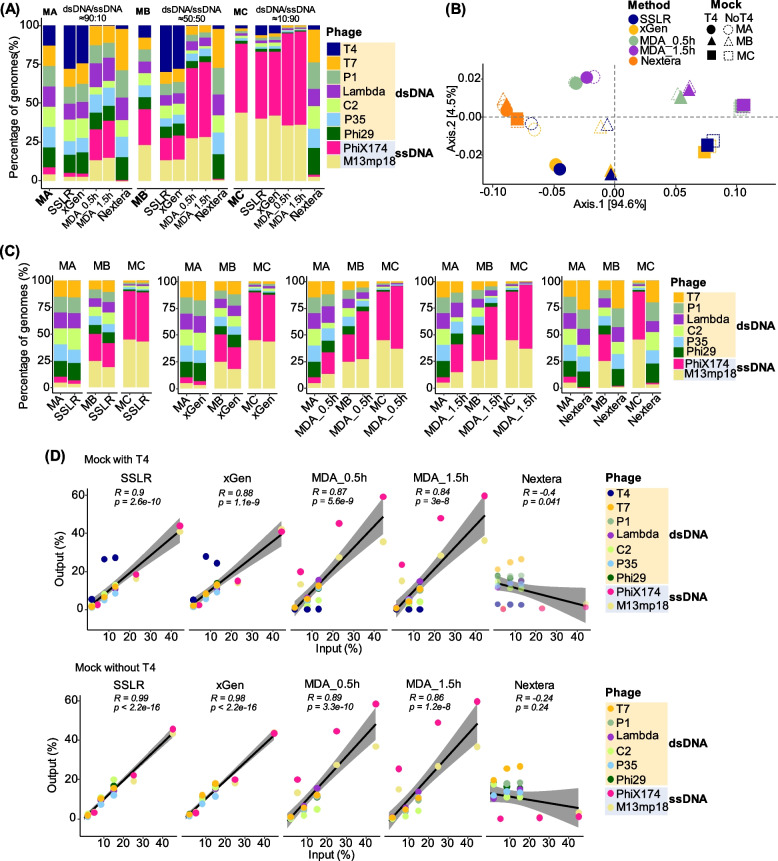
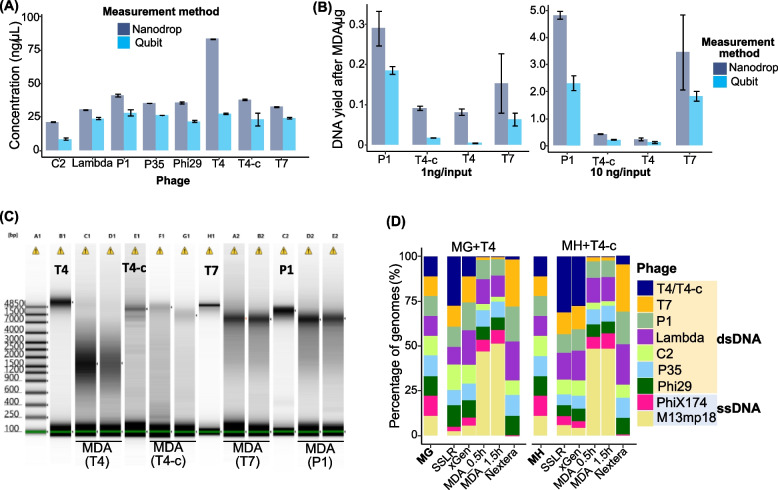
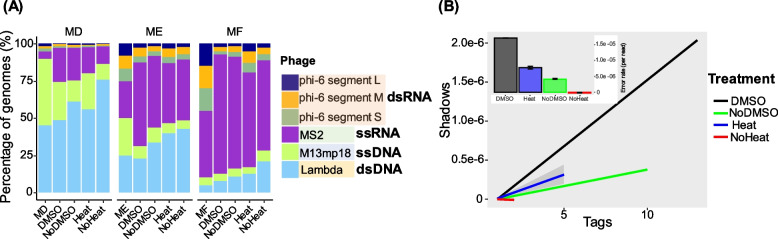
Results: We repurposed the ligation-based single-stranded library (SSLR) preparation method for virome studies. We demonstrate that the SSLR method exhibits exceptional efficiency in quantifying viral DNA genomes (both dsDNA and ssDNA) and outperforms existing double-stranded (Nextera) and single-stranded (xGen, MDA + Nextera) library preparation approaches in terms of minimal amplification bias, evenness of coverage, and integrity of assembling viral genomes. The SSLR method can be utilized for the simultaneous library preparation of both DNA and RNA viral genomes. Furthermore, the SSLR method showed its ability to capture highly modified phage genomes, which were often lost using other library preparation approaches.
Conclusion: We introduce and improve a fast, simple, and efficient ligation-based single-stranded DNA library preparation for gut virome study. This method is compatible with Illumina sequencing platforms and only requires ligation reagents within 3-h library preparation, which is similar or even better than the advanced library preparation method (xGen). We hope this method can be further optimized, validated, and widely used to make gut virome study more comparable and reproducible. Video Abstract.
Keywords: DsDNA virome; Gut virome; Modified nucleotides; Phage mock community; RNA virome; Single-stranded library; SsDNA virome.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
