

Apolipoprotein-L1 (APOL1): From Sleeping Sickness to Kidney Disease
- PMID: 39451256
- PMCID: PMC11506758
- DOI: 10.3390/cells13201738
Apolipoprotein-L1 (APOL1): From Sleeping Sickness to Kidney Disease
Abstract
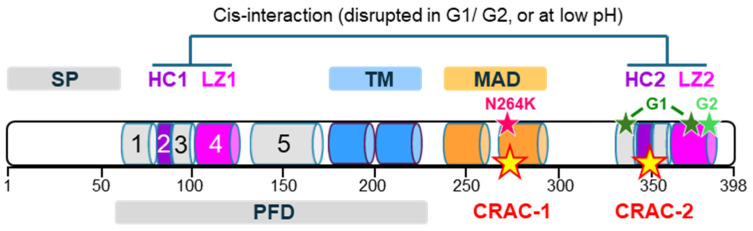
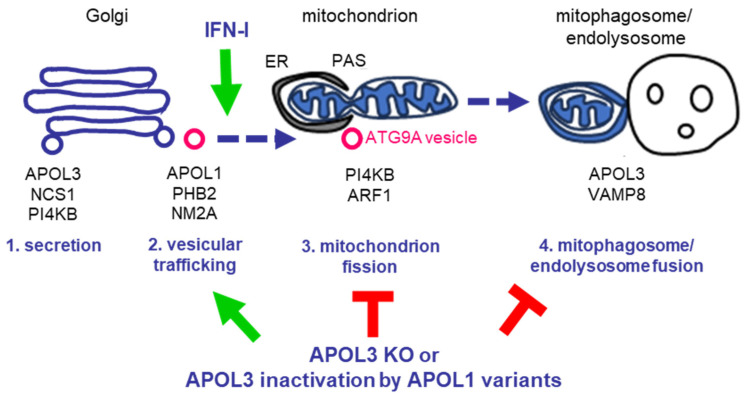
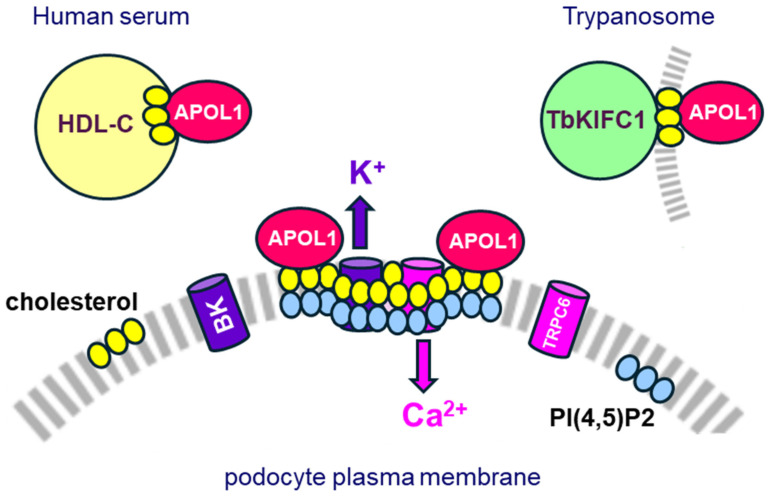
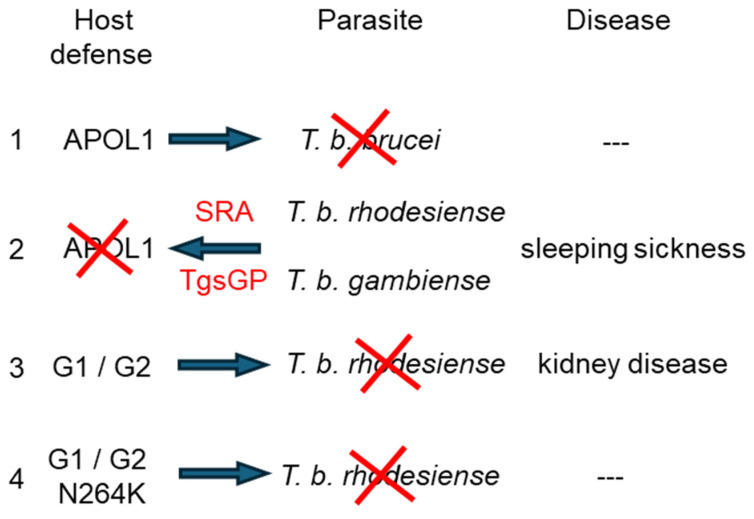
Apolipoprotein-L1 (APOL1) is a membrane-interacting protein induced by inflammation, which confers human resistance to infection by African trypanosomes. APOL1 kills Trypanosoma brucei through induction of apoptotic-like parasite death, but two T. brucei clones acquired resistance to APOL1, allowing them to cause sleeping sickness. An APOL1 C-terminal sequence alteration, such as occurs in natural West African variants G1 and G2, restored human resistance to these clones. However, APOL1 unfolding induced by G1 or G2 mutations enhances protein hydrophobicity, resulting in kidney podocyte dysfunctions affecting renal filtration. The mechanism involved in these dysfunctions is debated. The ability of APOL1 to generate ion pores in trypanosome intracellular membranes or in synthetic membranes was provided as an explanation. However, transmembrane insertion of APOL1 strictly depends on acidic conditions, and podocyte cytopathology mainly results from secreted APOL1 activity on the plasma membrane, which occurs under non-acidic conditions. In this review, I argue that besides inactivation of APOL3 functions in membrane dynamics (fission and fusion), APOL1 variants induce inflammation-linked podocyte toxicity not through pore formation, but through plasma membrane disturbance resulting from increased interaction with cholesterol, which enhances cation channels activity. A natural mutation in the membrane-interacting domain (N264K) abrogates variant APOL1 toxicity at the expense of slightly increased sensitivity to trypanosomes, further illustrating the continuous mutual adaptation between host and parasite.
Keywords: APOL1 risk variants; ARF1; INF-I inflammation; N264K variant; NCS1; PI4KB; apoptosis; cation channels; cholesterol microdomains; chronic kidney disease; membrane dynamics; mitochondrion fission; mitophagy; sleeping sickness.
Conflict of interest statement
The author declares no conflicts of interest.
Figures
References
-
- Duchateau P.N., Pullinger C.R., Orellana R.E., Kunitake S.T., Naya-Vigne J., O’Connor P.M., Malloy M.J., Kane J.P. Apolipoprotein L, a new human high density lipoprotein apolipoprotein expressed by the pancreas. Identification, cloning, characterization, and plasma distribution of apolipoprotein L. J. Biol. Chem. 1997;272:25576–25582. doi: 10.1074/jbc.272.41.25576. - DOI - PubMed
-
- Uzureau S., Coquerelle C., Vermeiren C., Uzureau P., Van Acker A., Pilotte L., Monteyne D., Acolty V., Vanhollebeke B., Van den Eynde B., et al. Apolipoproteins L control cell death triggered by TLR3/TRIF signaling in dendritic cells. Eur. J. Immunol. 2016;46:1854–1866. doi: 10.1002/eji.201546252. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
