

Aquificae overcomes competition by archaeal thermophiles, and crowding by bacterial mesophiles, to dominate the boiling vent-water of a Trans-Himalayan sulfur-borax spring
- PMID: 39453910
- PMCID: PMC11508158
- DOI: 10.1371/journal.pone.0310595
Aquificae overcomes competition by archaeal thermophiles, and crowding by bacterial mesophiles, to dominate the boiling vent-water of a Trans-Himalayan sulfur-borax spring
Abstract
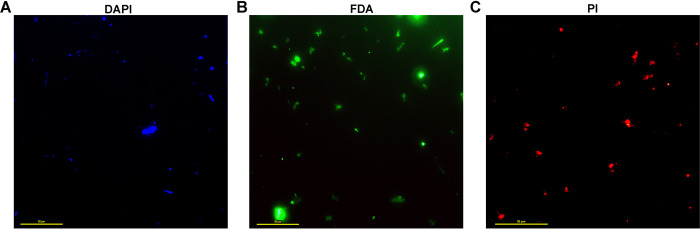
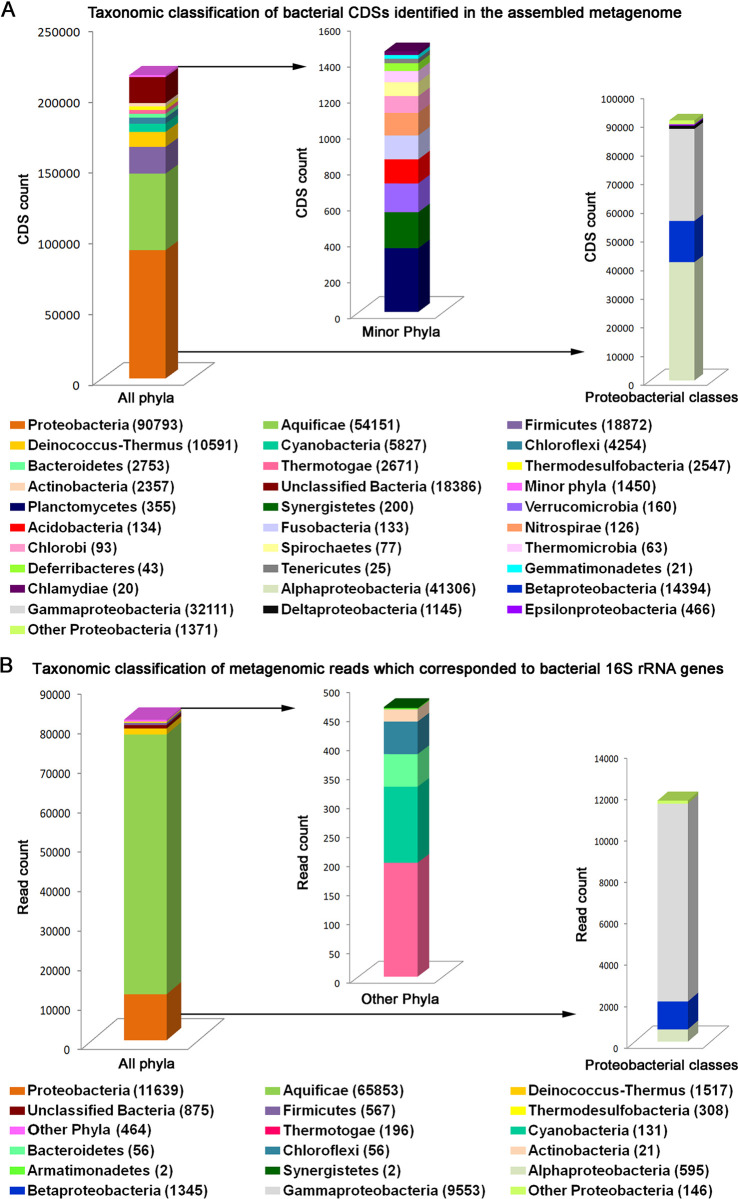
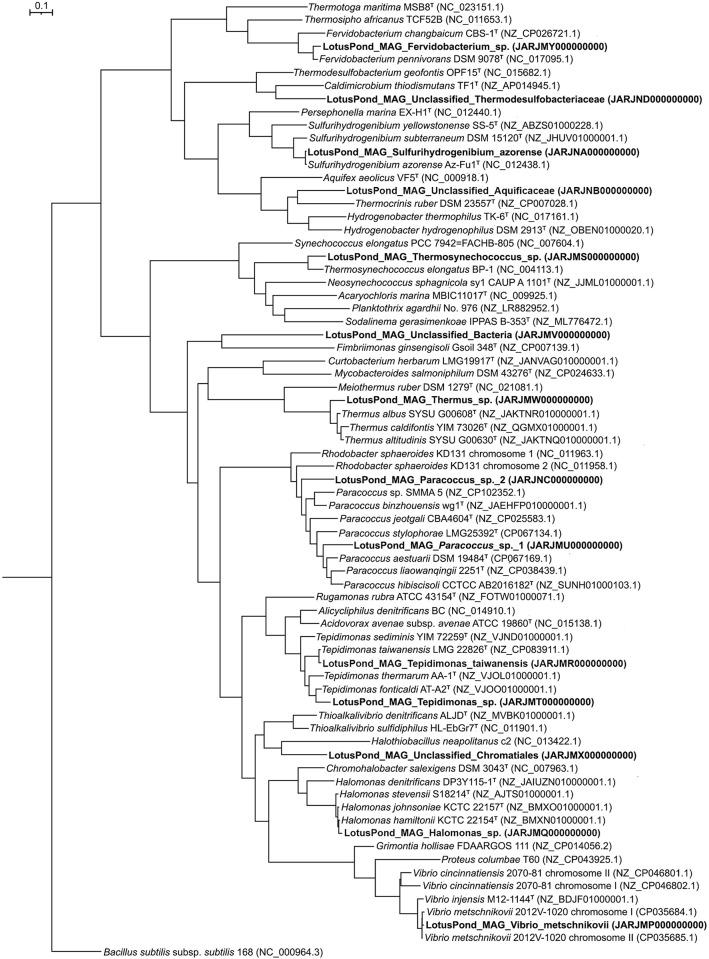
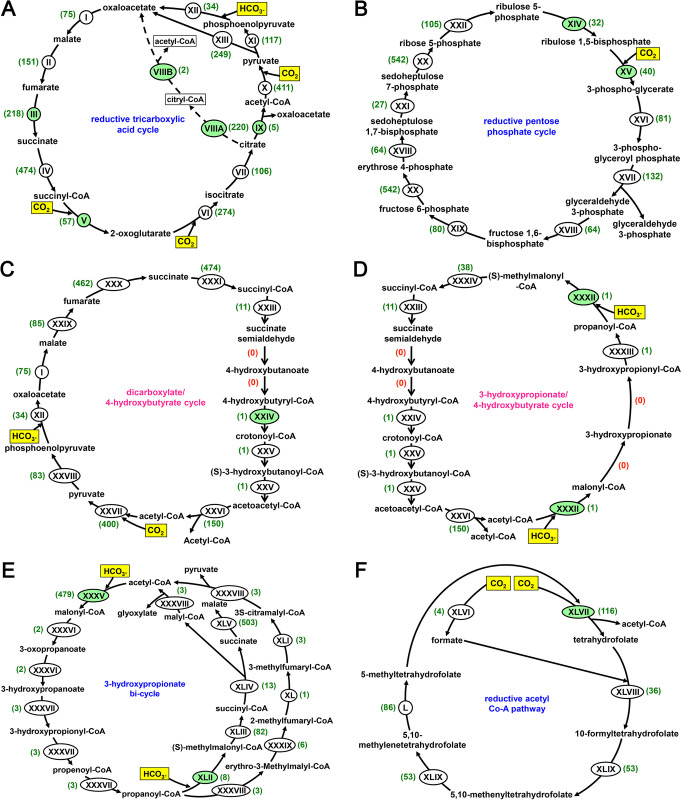
Trans-Himalayan hot spring waters rich in boron, chlorine, sodium and sulfur (but poor in calcium and silicon) are known based on PCR-amplified 16S rRNA gene sequence data to harbor high diversities of infiltrating bacterial mesophiles. Yet, little is known about the community structure and functions, primary productivity, mutual interactions, and thermal adaptations of the microorganisms present in the steaming waters discharged by these geochemically peculiar spring systems. We revealed these aspects of a bacteria-dominated microbiome (microbial cell density ~8.5 × 104 mL-1; live:dead cell ratio 1.7) thriving in the boiling (85°C) fluid vented by a sulfur-borax spring called Lotus Pond, situated at 4436 m above the mean sea-level, in the Puga valley of eastern Ladakh, on the Changthang plateau. Assembly, annotation, and population-binning of >15-GB metagenomic sequence illuminated the numeral predominance of Aquificae. While members of this phylum accounted for 80% of all 16S rRNA-encoding reads within the metagenomic dataset, 14% of such reads were attributed to Proteobacteria. Post assembly, only 25% of all protein-coding genes identified were attributable to Aquificae, whereas 41% was ascribed to Proteobacteria. Annotation of metagenomic reads encoding 16S rRNAs, and/or PCR-amplified 16S rRNA genes, identified 163 bacterial genera, out of which 66 had been detected in past investigations of Lotus Pond's vent-water via 16S amplicon sequencing. Among these 66, Fervidobacterium, Halomonas, Hydrogenobacter, Paracoccus, Sulfurihydrogenibium, Tepidimonas, Thermus and Thiofaba (or their close phylogenomic relatives) were presently detected as metagenome-assembled genomes (MAGs). Remarkably, the Hydrogenobacter related MAG alone accounted for ~56% of the entire metagenome, even though only 15 out of the 66 genera consistently present in Lotus Pond's vent-water have strains growing in the laboratory at >45°C, reflecting the continued existence of the mesophiles in the ecosystem. Furthermore, the metagenome was replete with genes crucial for thermal adaptation in the context of Lotus Pond's geochemistry and topography. In terms of sequence similarity, a majority of those genes were attributable to phylogenetic relatives of mesophilic bacteria, while functionally they rendered functions such as encoding heat shock proteins, molecular chaperones, and chaperonin complexes; proteins controlling/modulating/inhibiting DNA gyrase; universal stress proteins; methionine sulfoxide reductases; fatty acid desaturases; different toxin-antitoxin systems; enzymes protecting against oxidative damage; proteins conferring flagellar structure/function, chemotaxis, cell adhesion/aggregation, biofilm formation, and quorum sensing. The Lotus Pond Aquificae not only dominated the microbiome numerically but also acted potentially as the main primary producers of the ecosystem, with chemolithotrophic sulfur oxidation (Sox) being the fundamental bioenergetic mechanism, and reductive tricarboxylic acid (rTCA) cycle the predominant carbon fixation pathway. The Lotus Pond metagenome contained several genes directly or indirectly related to virulence functions, biosynthesis of secondary metabolites including antibiotics, antibiotic resistance, and multi-drug efflux pumping. A large proportion of these genes being attributable to Aquificae, and Proteobacteria (very few were ascribed to Archaea), it could be worth exploring in the future whether antibiosis helped the Aquificae overcome niche overlap with other thermophiles (especially those belonging to Archaea), besides exacerbating the bioenergetic costs of thermal endurance for the mesophilic intruders of the ecosystem.
Copyright: © 2024 Mondal et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

Similar articles
-
Genome-resolved metagenomics revealed metal-resistance, geochemical cycles in a Himalayan hot spring.Appl Microbiol Biotechnol. 2023 May;107(10):3273-3289. doi: 10.1007/s00253-023-12503-6. Epub 2023 Apr 13. Appl Microbiol Biotechnol. 2023. PMID: 37052633
-
Microbial diversity and biochemical potential encoded by thermal spring metagenomes derived from the Kamchatka Peninsula.Archaea. 2013;2013:136714. doi: 10.1155/2013/136714. Epub 2013 Feb 27. Archaea. 2013. PMID: 23533327 Free PMC article.
-
Bacteria and Archaea diversity within the hot springs of Lake Magadi and Little Magadi in Kenya.BMC Microbiol. 2016 Jul 7;16(1):136. doi: 10.1186/s12866-016-0748-x. BMC Microbiol. 2016. PMID: 27388368 Free PMC article.
-
Comparative metagenomic analyses of a high-altitude Himalayan geothermal spring revealed temperature-constrained habitat-specific microbial community and metabolic dynamics.Arch Microbiol. 2019 Apr;201(3):377-388. doi: 10.1007/s00203-018-01616-6. Epub 2019 Jan 25. Arch Microbiol. 2019. PMID: 30683956
-
How Do Thermophiles Organize Their Genomes?Microbes Environ. 2024;39(5):ME23087. doi: 10.1264/jsme2.ME23087. Microbes Environ. 2024. PMID: 38839371 Free PMC article. Review.
References
-
- Fields PA. Protein function at thermal extremes: balancing stability and flexibility. Comp Biochem Physiol A Mol Integr Physiol. 2001;129: 417–431. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
