

Advances in myocardial energy metabolism: metabolic remodelling in heart failure and beyond
- PMID: 39453987
- PMCID: PMC11646102
- DOI: 10.1093/cvr/cvae231
Advances in myocardial energy metabolism: metabolic remodelling in heart failure and beyond
Abstract
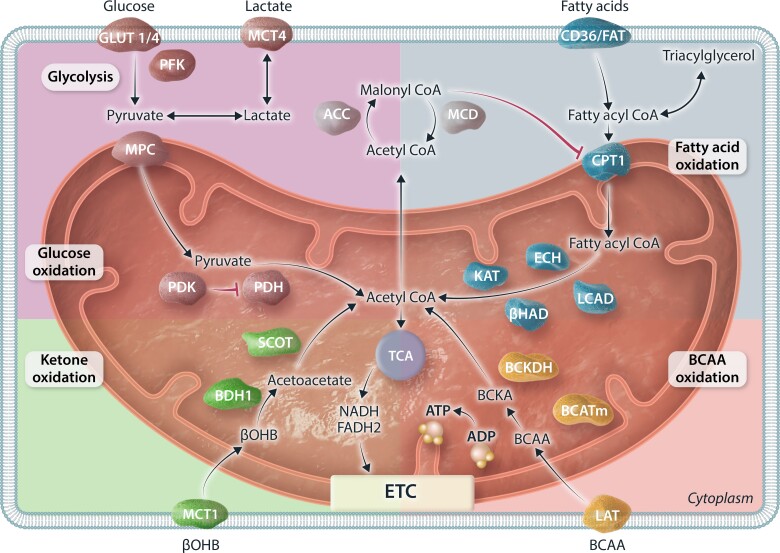
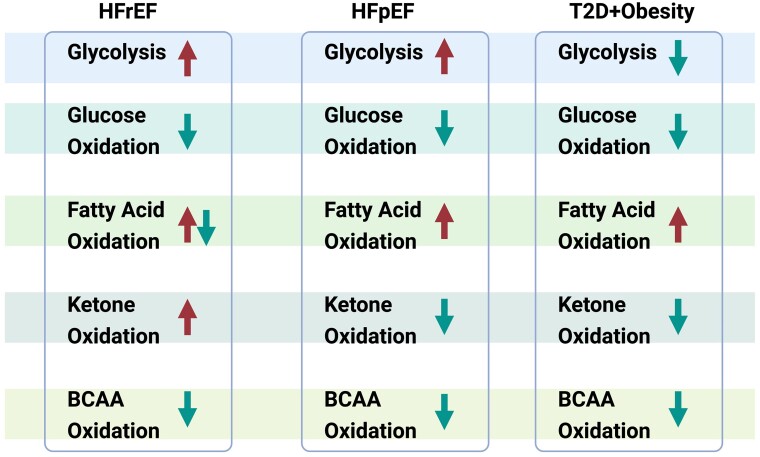
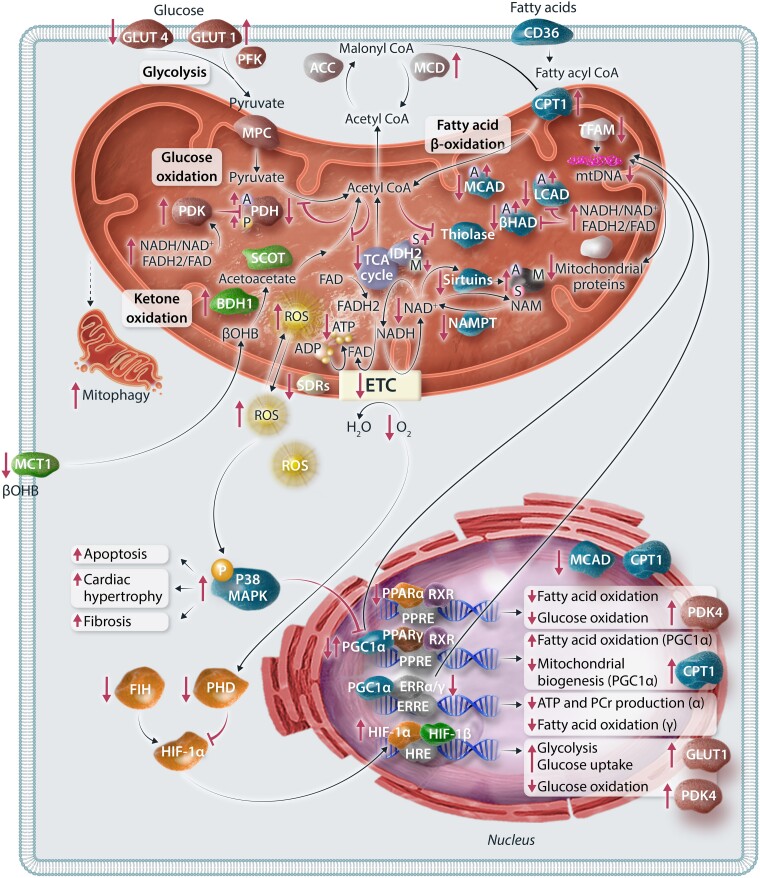
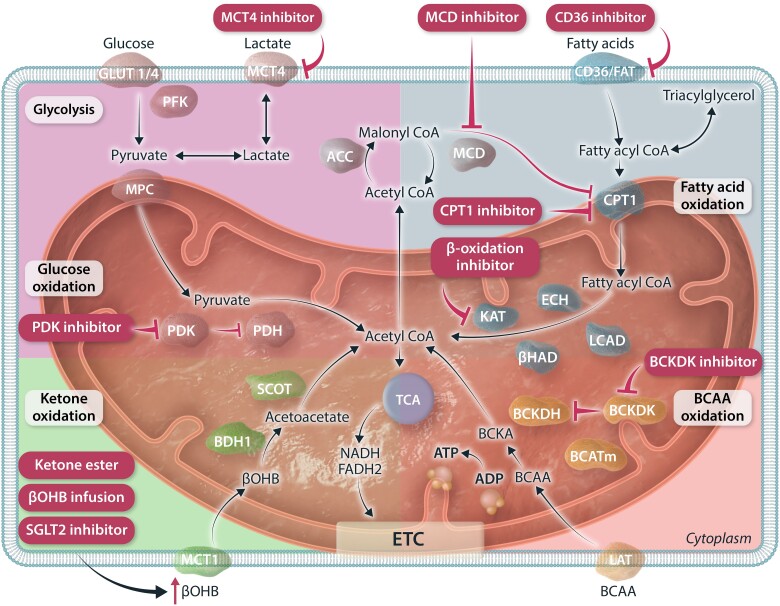
The very high energy demand of the heart is primarily met by adenosine triphosphate (ATP) production from mitochondrial oxidative phosphorylation, with glycolysis providing a smaller amount of ATP production. This ATP production is markedly altered in heart failure, primarily due to a decrease in mitochondrial oxidative metabolism. Although an increase in glycolytic ATP production partly compensates for the decrease in mitochondrial ATP production, the failing heart faces an energy deficit that contributes to the severity of contractile dysfunction. The relative contribution of the different fuels for mitochondrial ATP production dramatically changes in the failing heart, which depends to a large extent on the type of heart failure. A common metabolic defect in all forms of heart failure [including heart failure with reduced ejection fraction (HFrEF), heart failure with preserved EF (HFpEF), and diabetic cardiomyopathies] is a decrease in mitochondrial oxidation of pyruvate originating from glucose (i.e. glucose oxidation). This decrease in glucose oxidation occurs regardless of whether glycolysis is increased, resulting in an uncoupling of glycolysis from glucose oxidation that can decrease cardiac efficiency. The mitochondrial oxidation of fatty acids by the heart increases or decreases, depending on the type of heart failure. For instance, in HFpEF and diabetic cardiomyopathies myocardial fatty acid oxidation increases, while in HFrEF myocardial fatty acid oxidation either decreases or remains unchanged. The oxidation of ketones (which provides the failing heart with an important energy source) also differs depending on the type of heart failure, being increased in HFrEF, and decreased in HFpEF and diabetic cardiomyopathies. The alterations in mitochondrial oxidative metabolism and glycolysis in the failing heart are due to transcriptional changes in key enzymes involved in the metabolic pathways, as well as alterations in redox state, metabolic signalling and post-translational epigenetic changes in energy metabolic enzymes. Of importance, targeting the mitochondrial energy metabolic pathways has emerged as a novel therapeutic approach to improving cardiac function and cardiac efficiency in the failing heart.
Keywords: Fatty acid oxidation; Glucose oxidation; HFpEF; HFrEF; Ketone oxidation.
© The Author(s) 2024. Published by Oxford University Press on behalf of the European Society of Cardiology.
Conflict of interest statement
Conflict of interest: G.D.L. is a shareholder of Metabolic Modulators Research Ltd and has received grant support from Servier, Boehringer Ingelheim, Sanofi and REMED Biopharmaceuticals. The other authors have no additional conflicts of interest relevant to this article to declare.
Figures
Similar articles
-
Cardiac Energy Metabolism in Heart Failure.Circ Res. 2021 May 14;128(10):1487-1513. doi: 10.1161/CIRCRESAHA.121.318241. Epub 2021 May 13. Circ Res. 2021. PMID: 33983836 Free PMC article. Review.
-
Uncoupling of glycolysis from glucose oxidation accompanies the development of heart failure with preserved ejection fraction.Mol Med. 2018 Mar 15;24(1):3. doi: 10.1186/s10020-018-0005-x. Mol Med. 2018. PMID: 30134787 Free PMC article.
-
Mitochondrial fatty acid oxidation is the major source of cardiac adenosine triphosphate production in heart failure with preserved ejection fraction.Cardiovasc Res. 2024 Mar 30;120(4):360-371. doi: 10.1093/cvr/cvae006. Cardiovasc Res. 2024. PMID: 38193548
-
Myocardial Energy Substrate Metabolism in Heart Failure : from Pathways to Therapeutic Targets.Curr Pharm Des. 2015;21(25):3654-64. doi: 10.2174/1381612821666150710150445. Curr Pharm Des. 2015. PMID: 26166604 Review.
-
Complex Energy Metabolic Changes in Heart Failure With Preserved Ejection Fraction and Heart Failure With Reduced Ejection Fraction.Can J Cardiol. 2017 Jul;33(7):860-871. doi: 10.1016/j.cjca.2017.03.009. Epub 2017 Mar 19. Can J Cardiol. 2017. PMID: 28579160 Review.
Cited by
-
Pediatric heart failure: Current approach and treatment.JHLT Open. 2025 Jun 27;9:100331. doi: 10.1016/j.jhlto.2025.100331. eCollection 2025 Aug. JHLT Open. 2025. PMID: 40697906 Free PMC article.
-
BCAA catabolism targeted therapy for heart failure with preserved ejection fraction.Theranostics. 2025 May 24;15(13):6257-6273. doi: 10.7150/thno.105894. eCollection 2025. Theranostics. 2025. PMID: 40521197 Free PMC article.
-
The ketone body 3-hydroxybutyrate increases cardiac output and cardiac contractility in a porcine model of cardiogenic shock: a randomized, blinded, crossover trial.Basic Res Cardiol. 2025 Jun;120(3):579-596. doi: 10.1007/s00395-025-01103-2. Epub 2025 Apr 12. Basic Res Cardiol. 2025. PMID: 40220139 Free PMC article.
-
Integrating multi-omics and machine learning strategies to explore the "gene-protein-metabolite" network in ischemic heart failure with Qi deficiency and blood stasis syndrome.Chin Med. 2025 Jul 17;20(1):93. doi: 10.1186/s13020-025-01151-9. Chin Med. 2025. PMID: 40671145 Free PMC article.
-
Coronary microvascular disease in heart failure with preserved ejection fraction.Physiol Rep. 2025 Aug;13(16):e70521. doi: 10.14814/phy2.70521. Physiol Rep. 2025. PMID: 40834289 Free PMC article. Review.
References
-
- Lüscher TF. Heart failure subgroups: HFrEF, HFmrEF, and HFpEF with or without mitral regurgitation. Eur Heart J 2018;39:1–4. - PubMed
-
- Ponikowski P, Voors A, Anker S, Bueno H, Cleland J, Coats A, Falk V, González-Juanatey J, Harjola V, Jankowska E, Authors/Task Force Members . 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: the task force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J 2016;37:2129–2200. - PubMed
-
- Steinberg BA, Zhao X, Heidenreich PA, Peterson ED, Bhatt DL, Cannon CP, Hernandez AF, Fonarow GC. Trends in patients hospitalized with heart failure and preserved left ventricular ejection fraction: prevalence, therapies, and outcomes. Circulation 2012;126:65–75. - PubMed
-
- Neubauer S. The failing heart — an engine out of fuel. N Engl J Med 2007;356:1140–1151. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
