

Autoregulation ensures vertical transmission of the linear prophage GIL01
- PMID: 39455843
- PMCID: PMC11511902
- DOI: 10.1038/s42003-024-07082-9
Autoregulation ensures vertical transmission of the linear prophage GIL01
Abstract
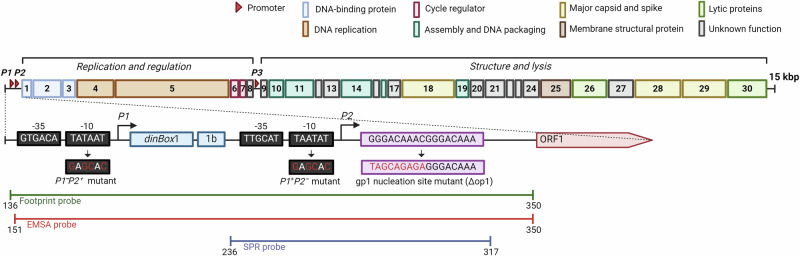
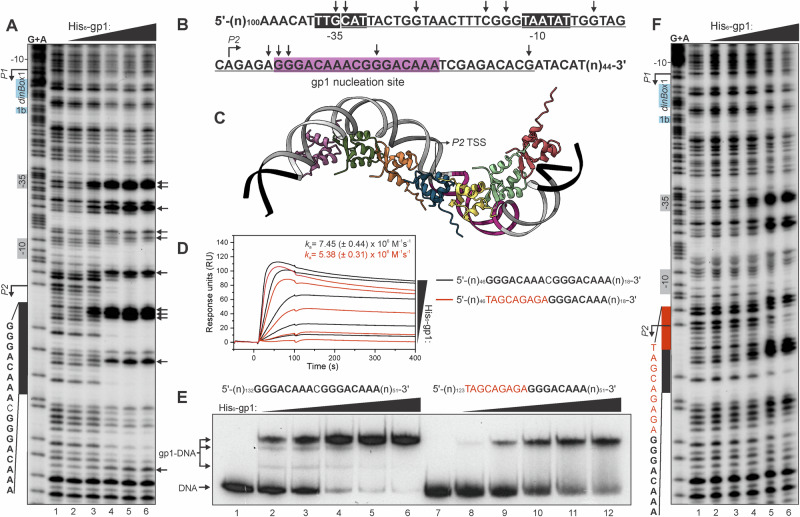
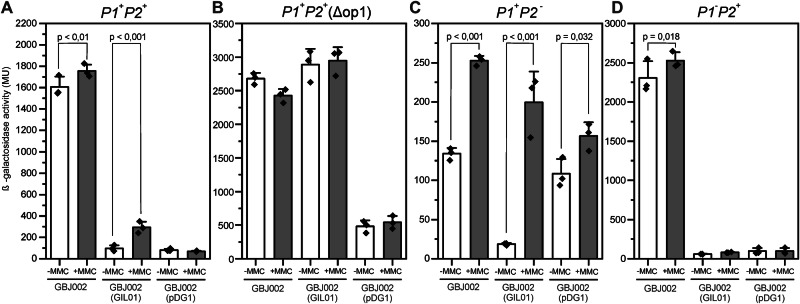
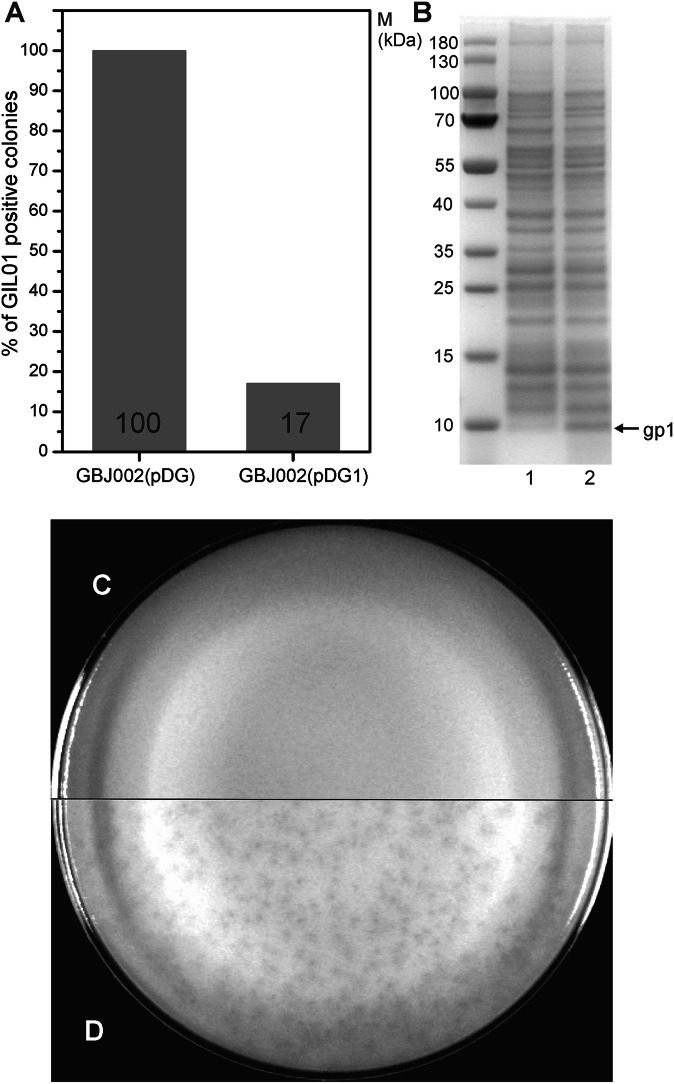
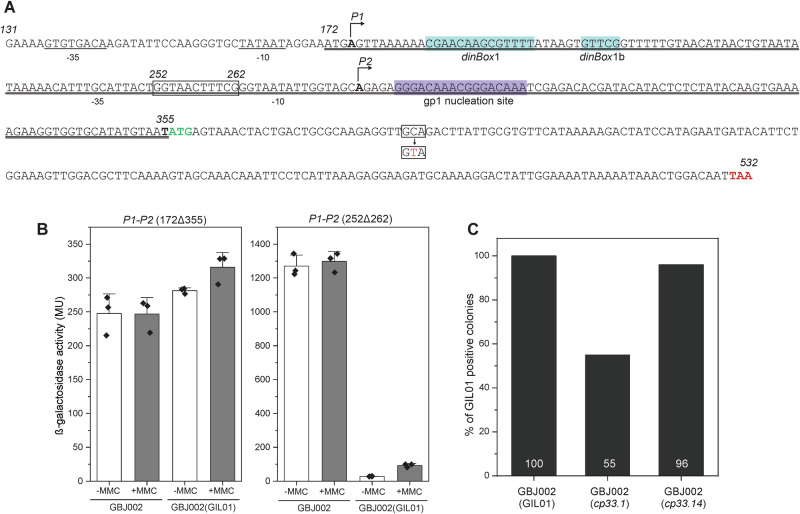
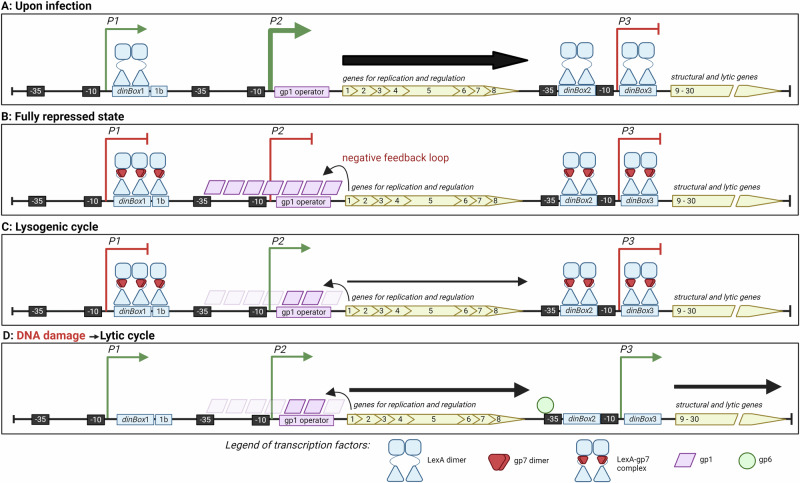
Betatectiviruses are prophages consisting of linear extrachromosomal genomes without obvious plasmid modules. It remains unclear how betatectiviruses are maintained in low-copy numbers in host cells and how they are vertically transmitted. Phage GIL01 is a model betatectivirus that infects the mosquito pathogen Bacillus thuringiensis serovar israelensis. Previous studies identified two closely spaced promoters, P1 and P2, responsible for the expression of GIL01 genes required for prophage replication and the switch from the lysogenic to lytic cycle. Here, we report that the GIL01-encoded 58-amino acid long gp1 protein forms a large nucleoprotein complex that represses its transcription from the strong promoter P2. Notably, ectopic expression of gp1 resulted in the loss of GIL01 in exponential cultures and immunized cells against infection with GIL01, indicating that gp1 plays a repressive role in the phage cycle. This finding is consistent with mutations in gp1 committing GIL01 to the lytic cycle and we show that maintenance of this phage variant in the bacterial population is contingent on the accumulation of deletions in the P1-P2 region. The fact that gp1 is conserved across most sequenced betatectiviruses suggests that the regulatory mechanism of gp1 that controls prophage maintenance is widespread among these bacteriophages.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
