

Public evidence on AI products for digital pathology
- PMID: 39455883
- PMCID: PMC11511888
- DOI: 10.1038/s41746-024-01294-3
Public evidence on AI products for digital pathology
Abstract
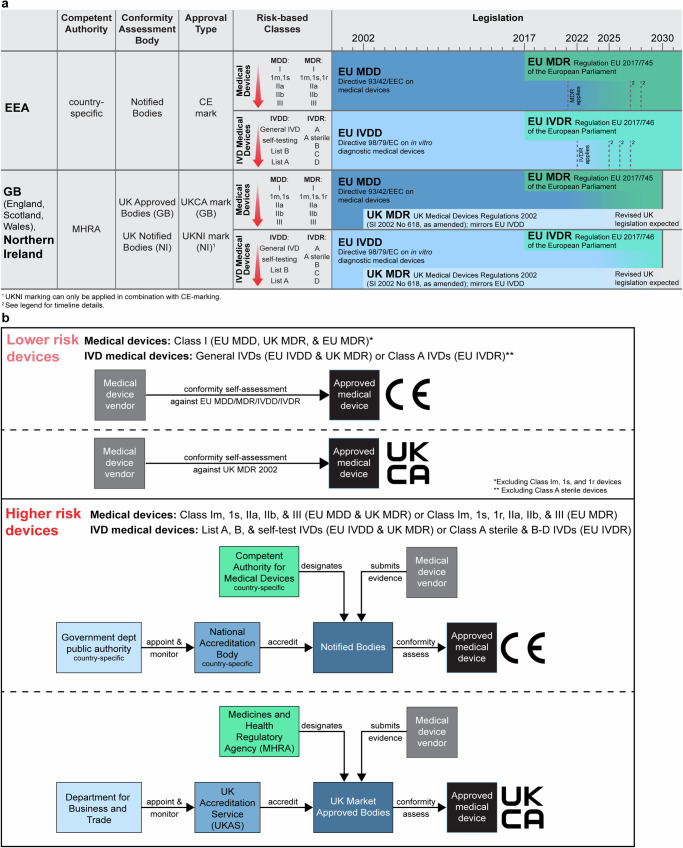
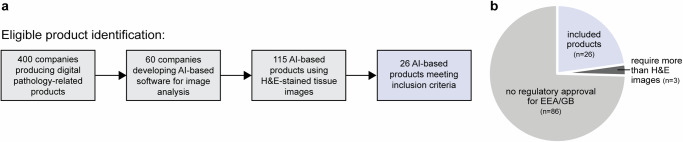
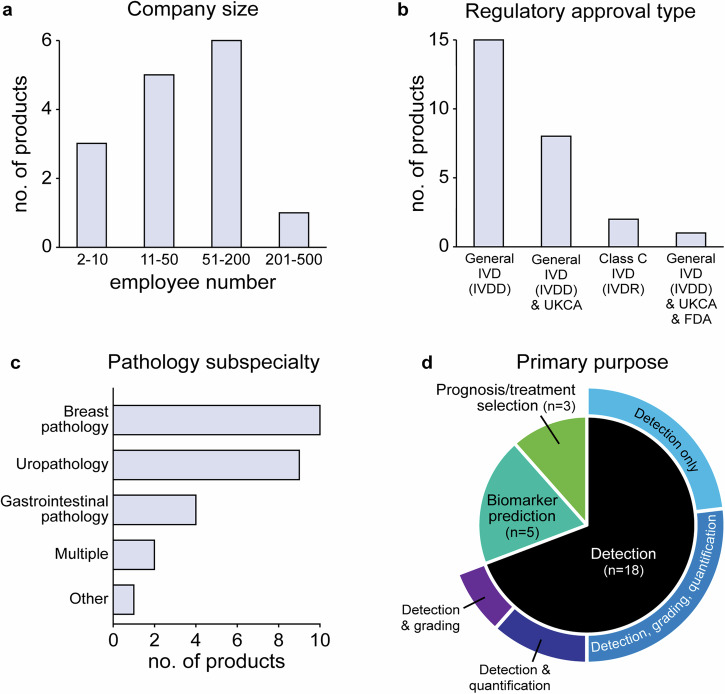
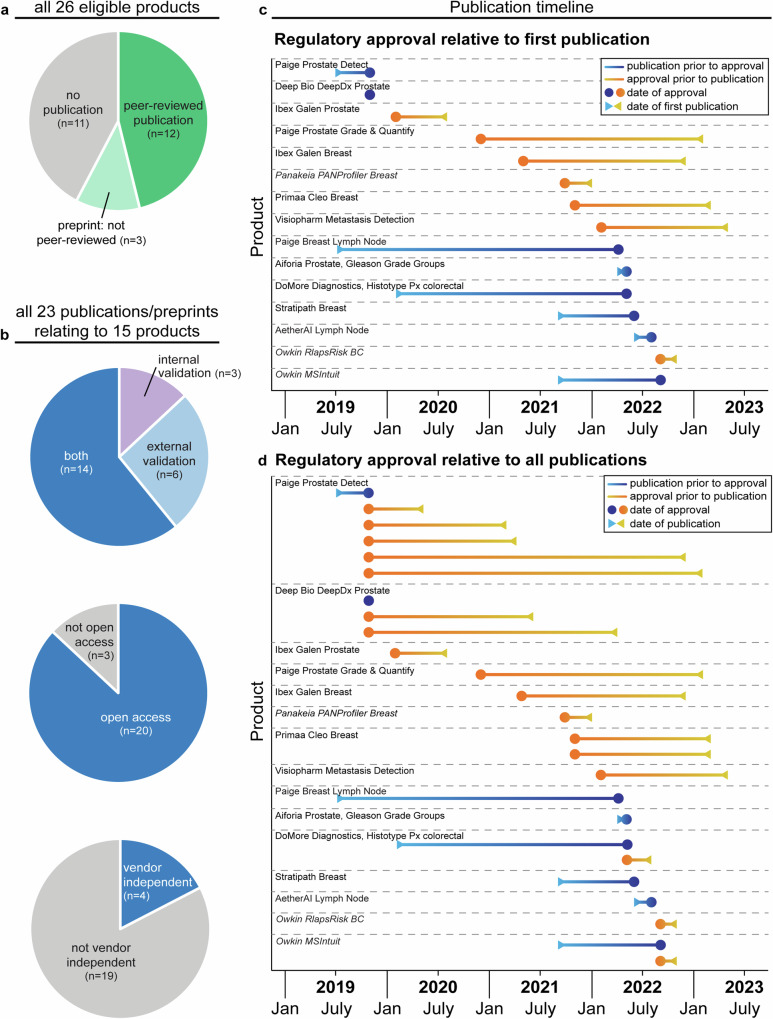
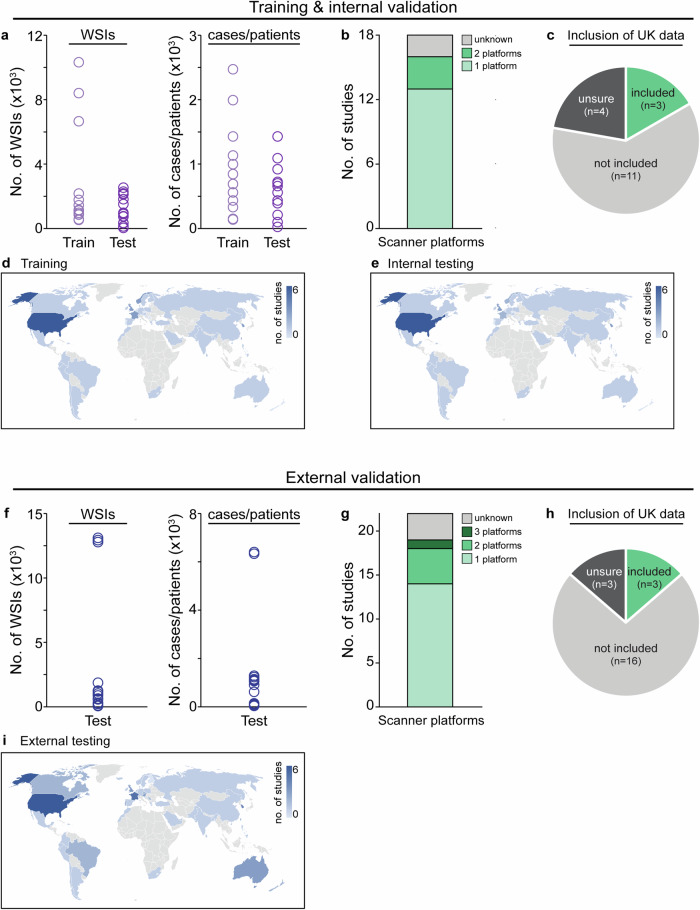
Novel products applying artificial intelligence (AI)-based methods to digital pathology images are touted to have many uses and benefits. However, publicly available information for products can be variable, with few sources of independent evidence. This review aimed to identify public evidence for AI-based products for digital pathology. Key features of products on the European Economic Area/Great Britain (EEA/GB) markets were examined, including their regulatory approval, intended use, and published validation studies. There were 26 AI-based products that met the inclusion criteria and, of these, 24 had received regulatory approval via the self-certification route as General in vitro diagnostic (IVD) medical devices. Only 10 of the products (38%) had peer-reviewed internal validation studies and 11 products (42%) had peer-reviewed external validation studies. To support transparency an online register was developed using identified public evidence ( https://osf.io/gb84r/ ), which we anticipate will provide an accessible resource on novel devices and support decision making.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Evidence standards framework (ESF) for digital health technologies. NICE, https://www.nice.org.uk/about/what-we-do/our-programmes/evidence-standar... (2023).
-
- Digital Technology Assessment Criteria (DTAC). NHS Transformation Directorate, https://transform.england.nhs.uk/key-tools-and-info/digital-technology-a... (2023).
-
- A guide to good practice for digital and data-driven health technologies. GOV.UK, https://www.gov.uk/government/publications/code-of-conduct-for-data-driv... (2021).
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
