

Effects of Angiotensin-I-Converting Enzyme (ACE) Mutations Associated with Alzheimer's Disease on Blood ACE Phenotype
- PMID: 39457722
- PMCID: PMC11504702
- DOI: 10.3390/biomedicines12102410
Effects of Angiotensin-I-Converting Enzyme (ACE) Mutations Associated with Alzheimer's Disease on Blood ACE Phenotype
Abstract
Backgrounds: Our recent analysis of 1200+ existing missense ACE mutations revealed that 400+ mutations are damaging and led us to hypothesize that carriers of heterozygous loss-of-function (LoF) ACE mutations (which result in low ACE levels) could be at risk for the development of late-onset Alzheimer's disease (AD).
Methods: Here, we quantified blood ACE levels in EDTA plasma from 41 subjects with 10 different heterozygous ACE mutations, as well as 33 controls, and estimated the effect of these mutations on ACE phenotype using a set of mAbs to ACE and two ACE substrates.
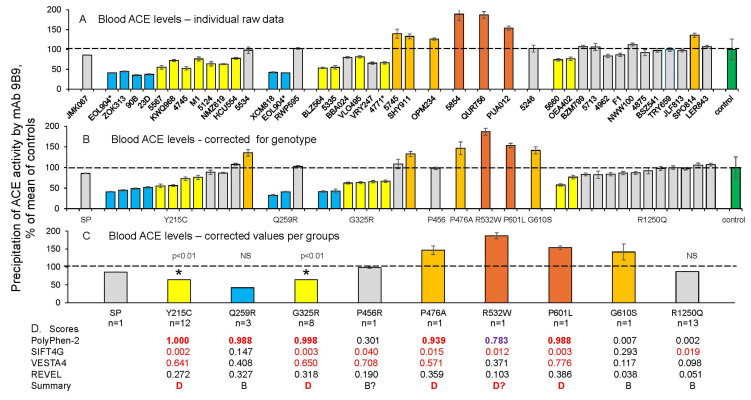
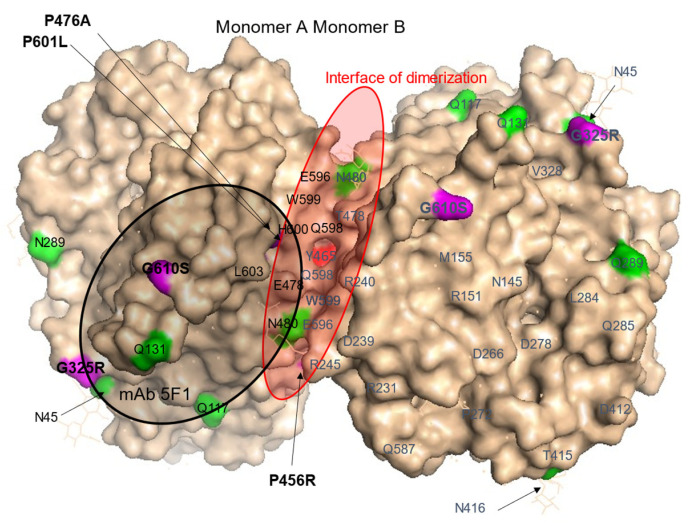
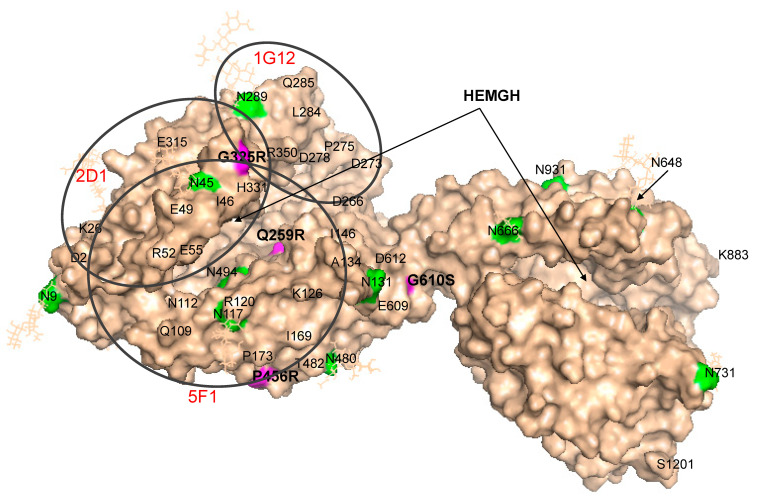
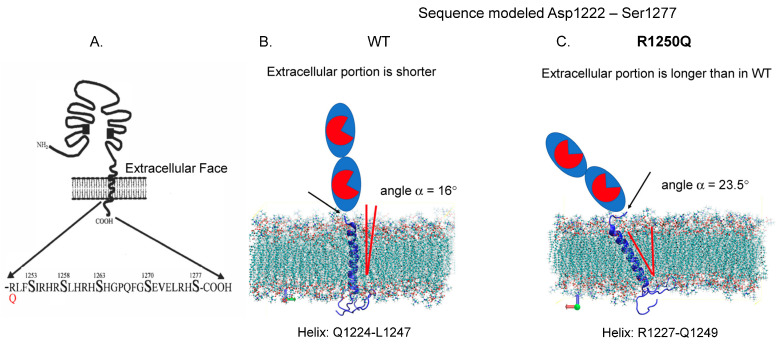
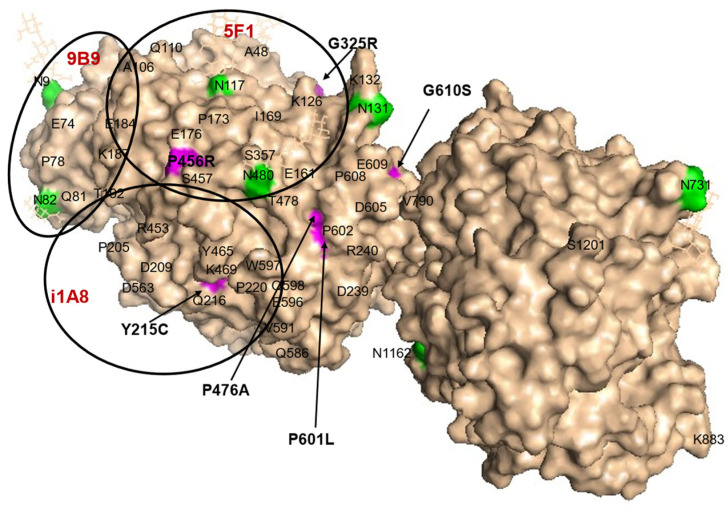
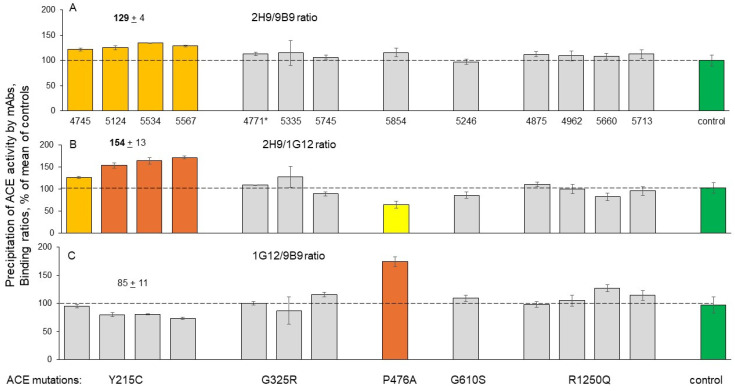
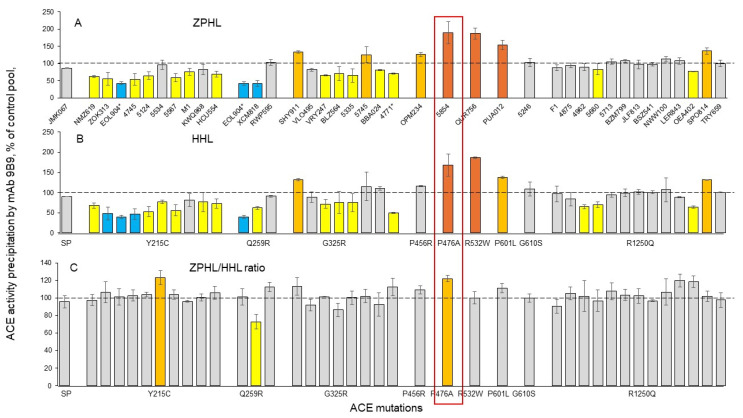
Results: We found that relatively frequent (~1%) AD-associated ACE mutations in the N domain of ACE, Y215C, and G325R are truly damaging and likely transport-deficient, with the ACE levels in plasma at only ~50% of controls. Another AD-associated ACE mutation, R1250Q, in the cytoplasmic tail, did not cause a decrease in ACE and likely did not affect surface ACE expression. We have also developed a method to identify patients with anti-catalytic mutations in the N domain. These mutations may result in reduced degradation of amyloid beta peptide Aβ42, an important component for amyloid deposition. Consequently, these could pose a risk factor for the development of AD.
Conclusions: Therefore, a systematic analysis of blood ACE levels in patients with all ACE mutations has the potential to identify individuals at an increased risk of late-onset AD. These individuals may benefit from future preventive or therapeutic interventions involving a combination of chemical and pharmacological chaperones, as well as proteasome inhibitors, aiming to enhance ACE protein traffic. This approach has been previously demonstrated in our cell model of the transport-deficient ACE mutation Q1069R.
Keywords: Alzheimer’s disease; angiotensin-I-converting enzyme; blood ACE; conformational changes; mutations; screening.
Conflict of interest statement
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Figures
References
-
- Andrade-Guerrero J., Santiago-Balmaseda A., Jeronimo-Aguilar P., Vargas-Rodríguez I., Cadena-Suárez A.R., Sánchez-Garibay C., Pozo-Molina G., Méndez-Catalá C.F., Cardenas-Aguayo M.D.C., Diaz-Cintra S., et al. Alzheimer’s Disease: An Updated Overview of Its Genetics. Int. J. Mol. Sci. 2023;24:3754. doi: 10.3390/ijms24043754. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous
